
Longitudinal Metabolomics Data Analysis Informed by Mechanistic Models
 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Real Meal Challenge Test Data
2.2. Simulated Meal Challenge Test Data
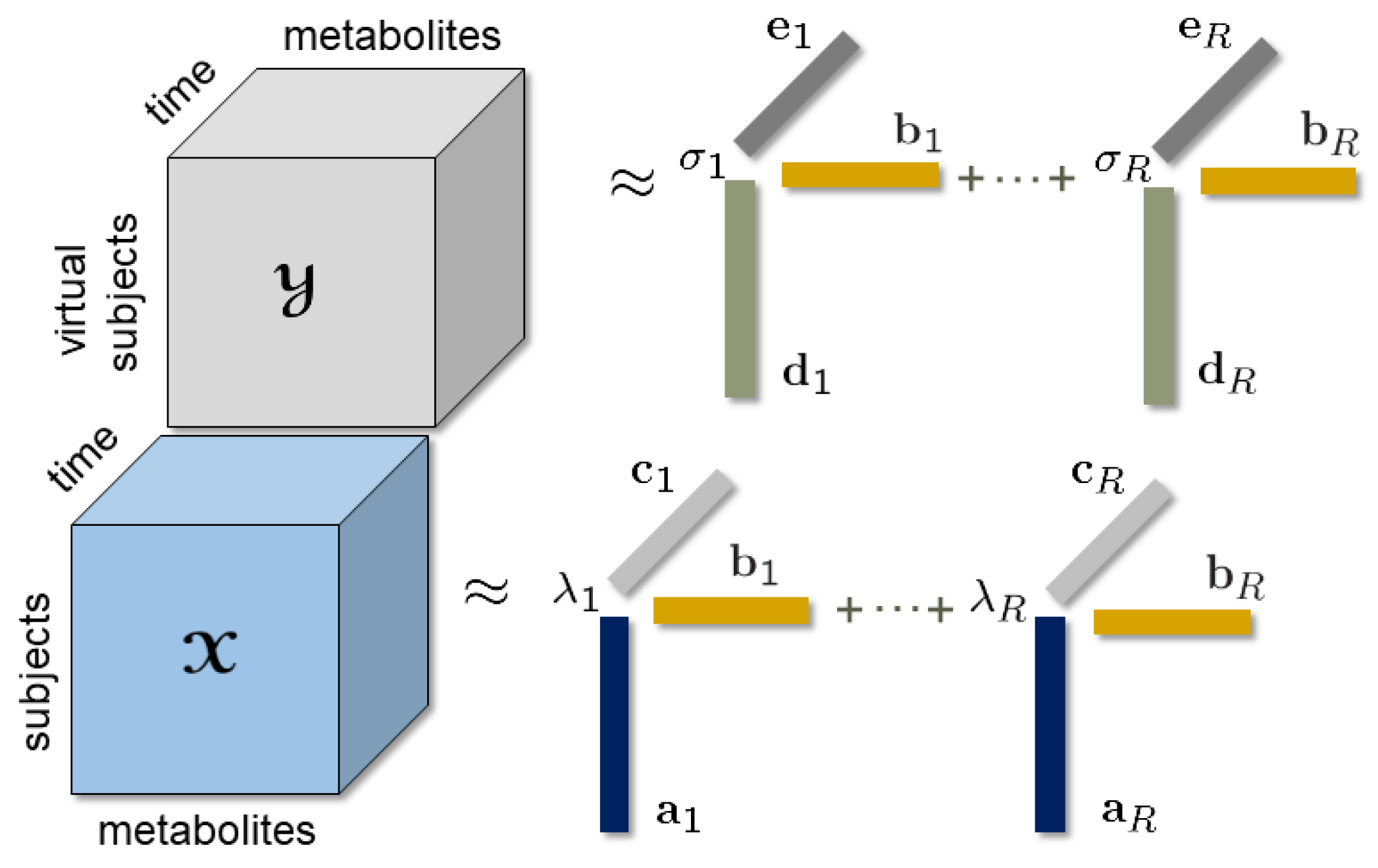
2.3. Tensor Factorizations
2.4. Coupled Tensor Factorizations
2.5. Experimental Set-Up
2.5.1. Data Preprocessing
2.5.2. Implementation Details
2.5.3. Model Selection
3. Results
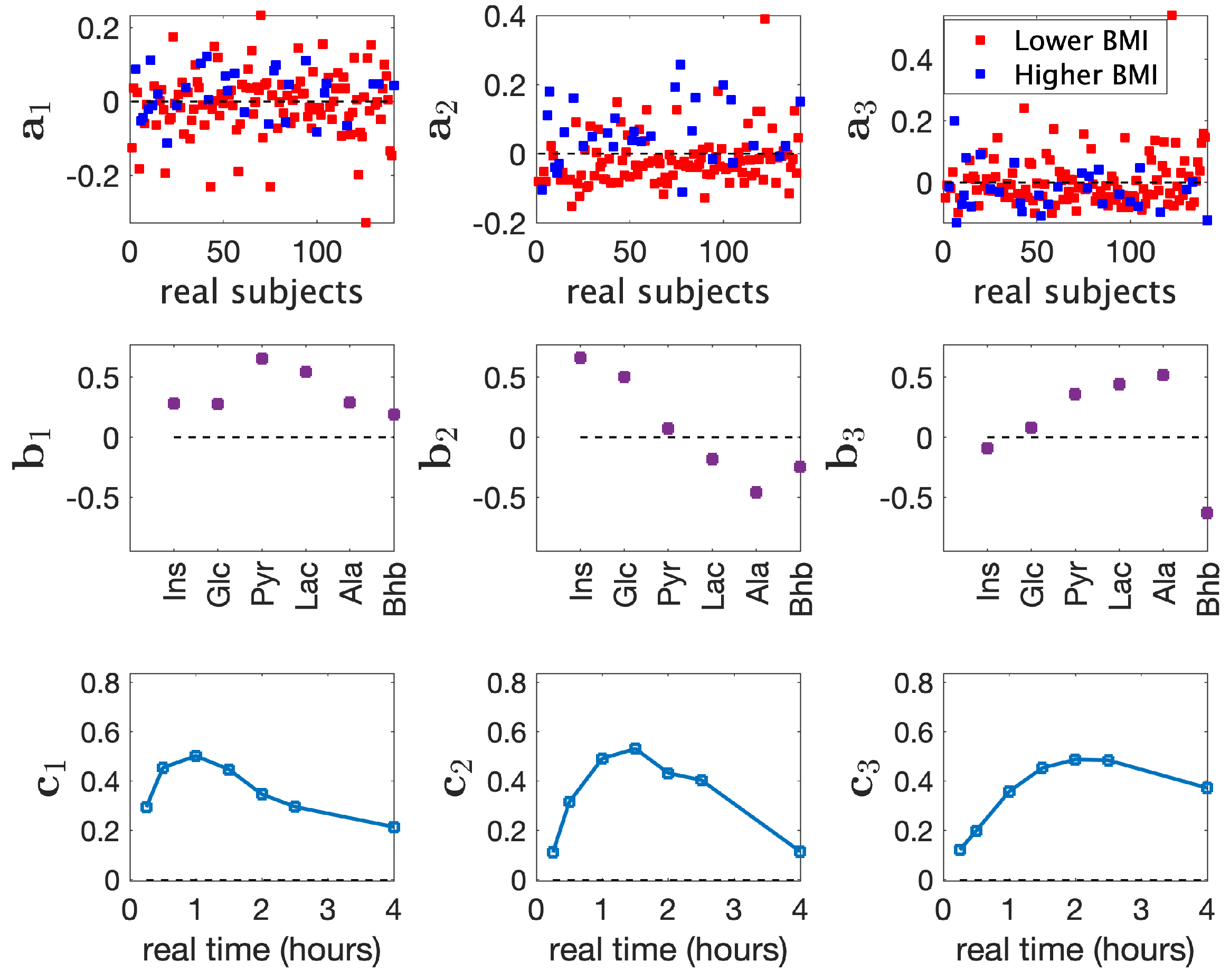
3.1. Analysis of Real Metabolomics Data
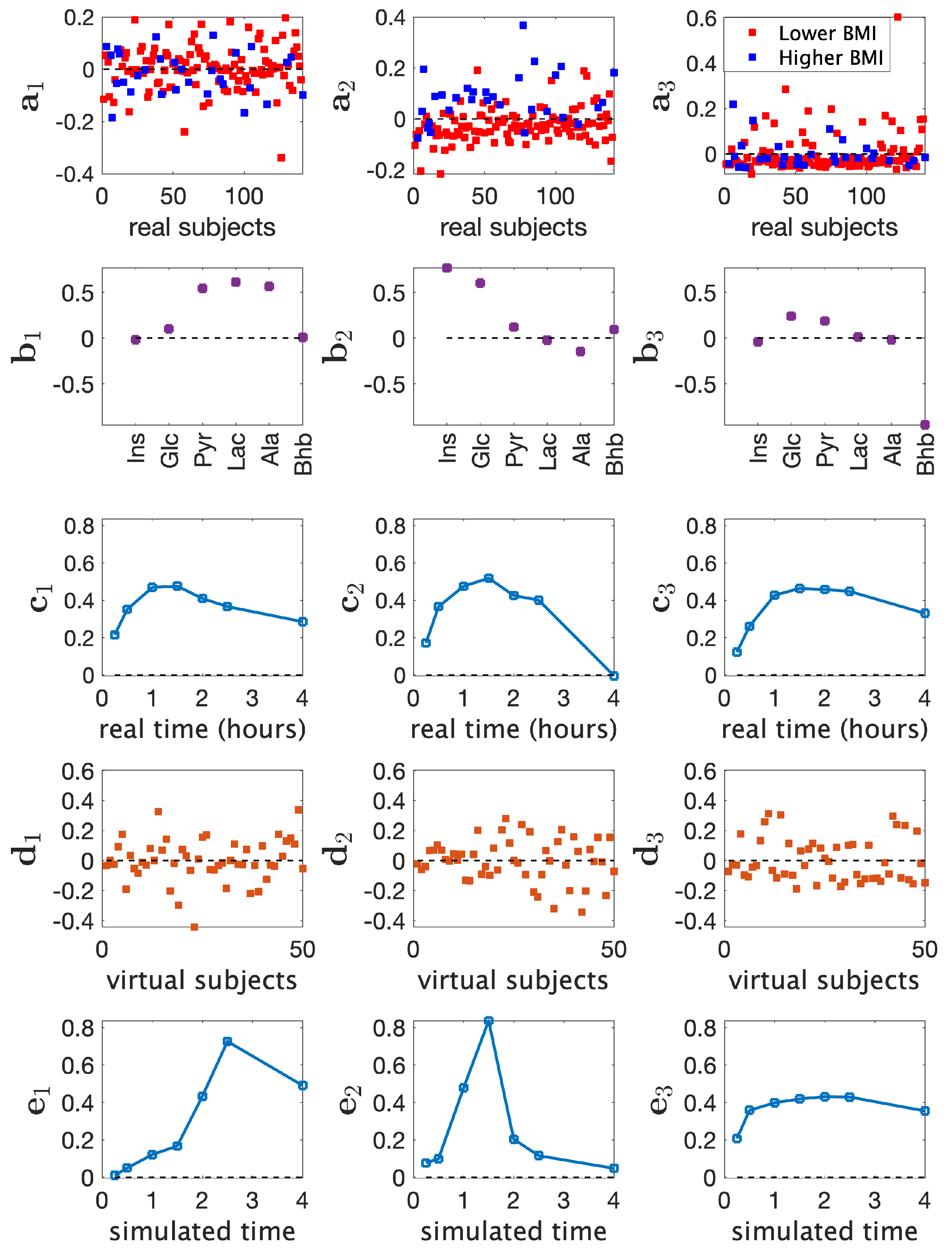
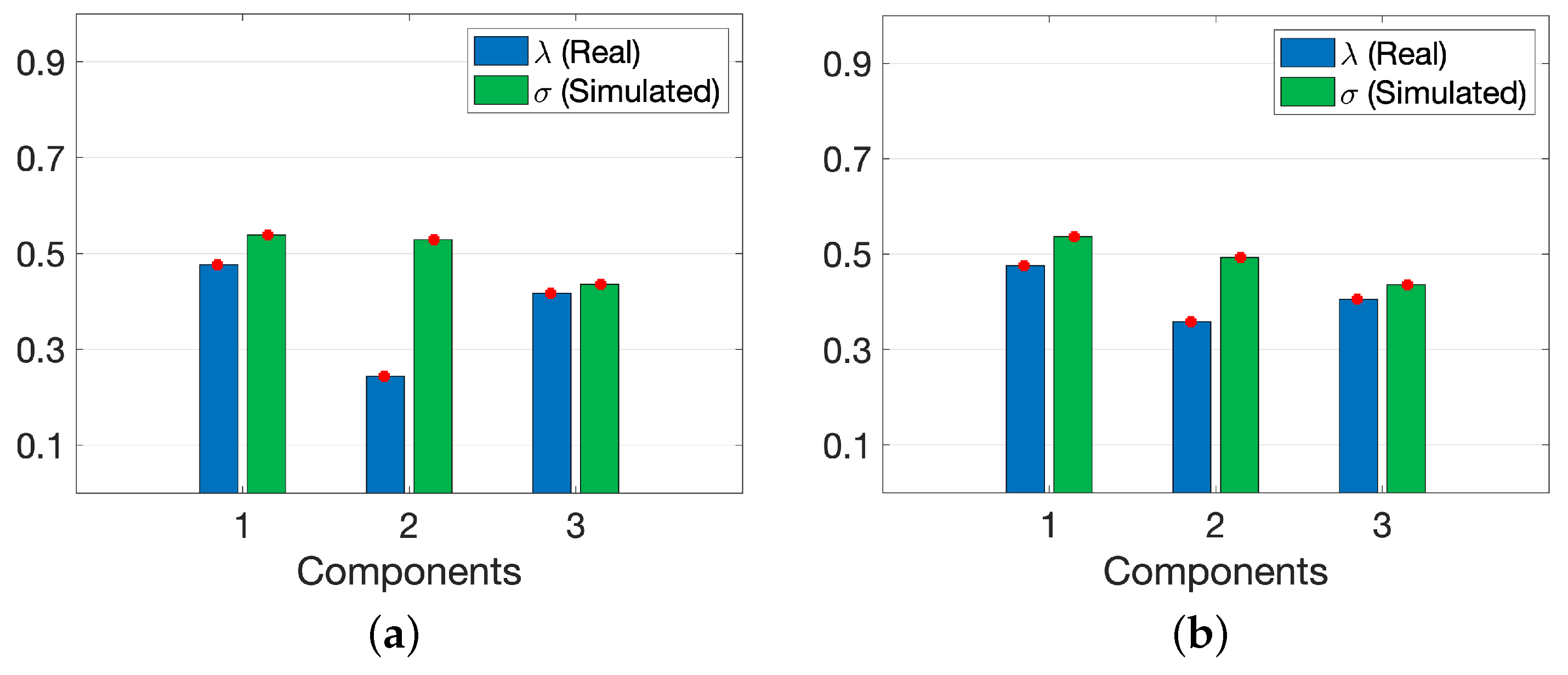
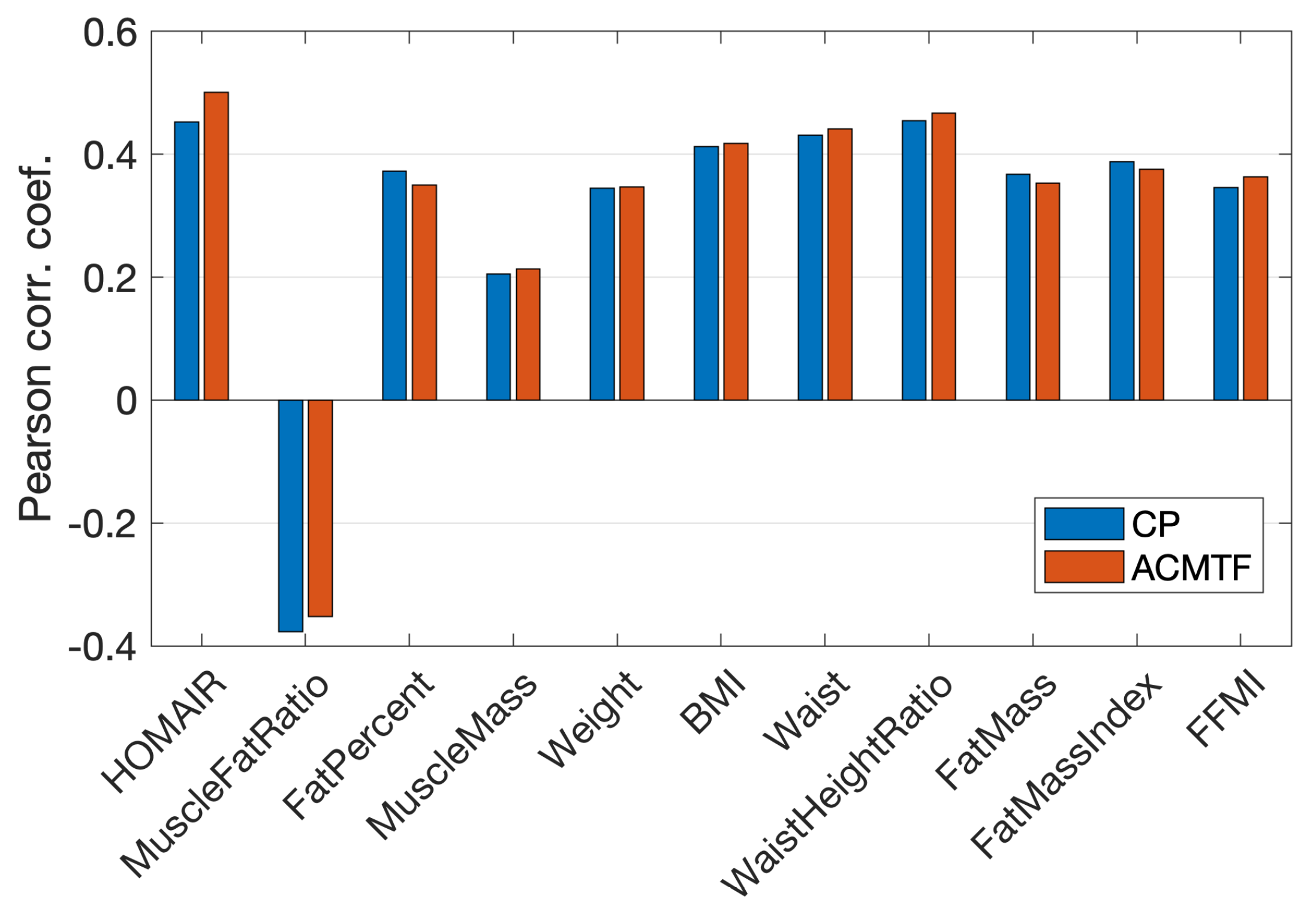
3.2. Joint Analysis of Real and Simulated Metabolomics Data
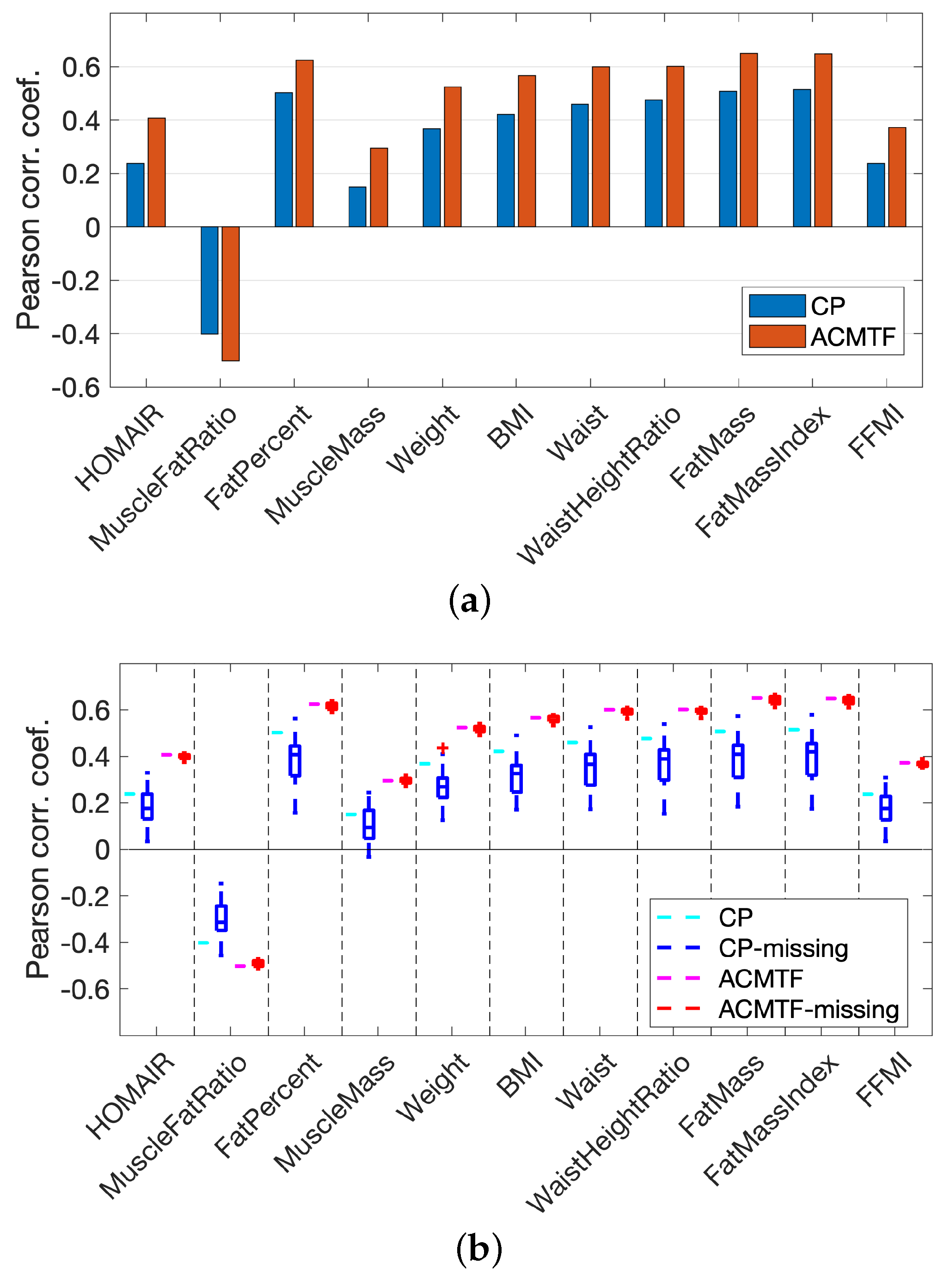
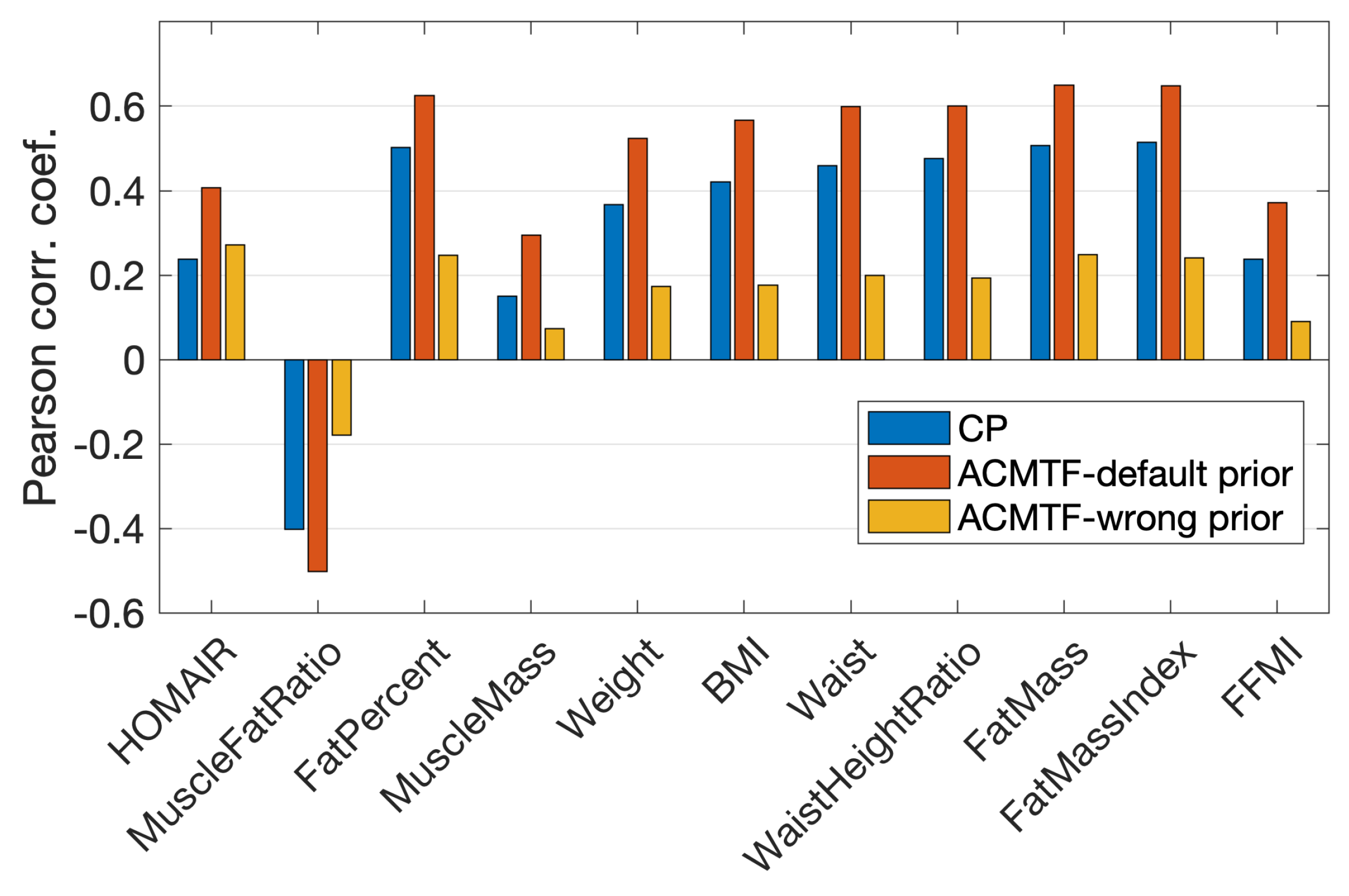
3.3. Analysis of Real Data vs. Joint Analysis of Simulated and Real Data
3.4. Joint Analysis of Real and Simulated Metabolomics Data in the Presence of Missing Data
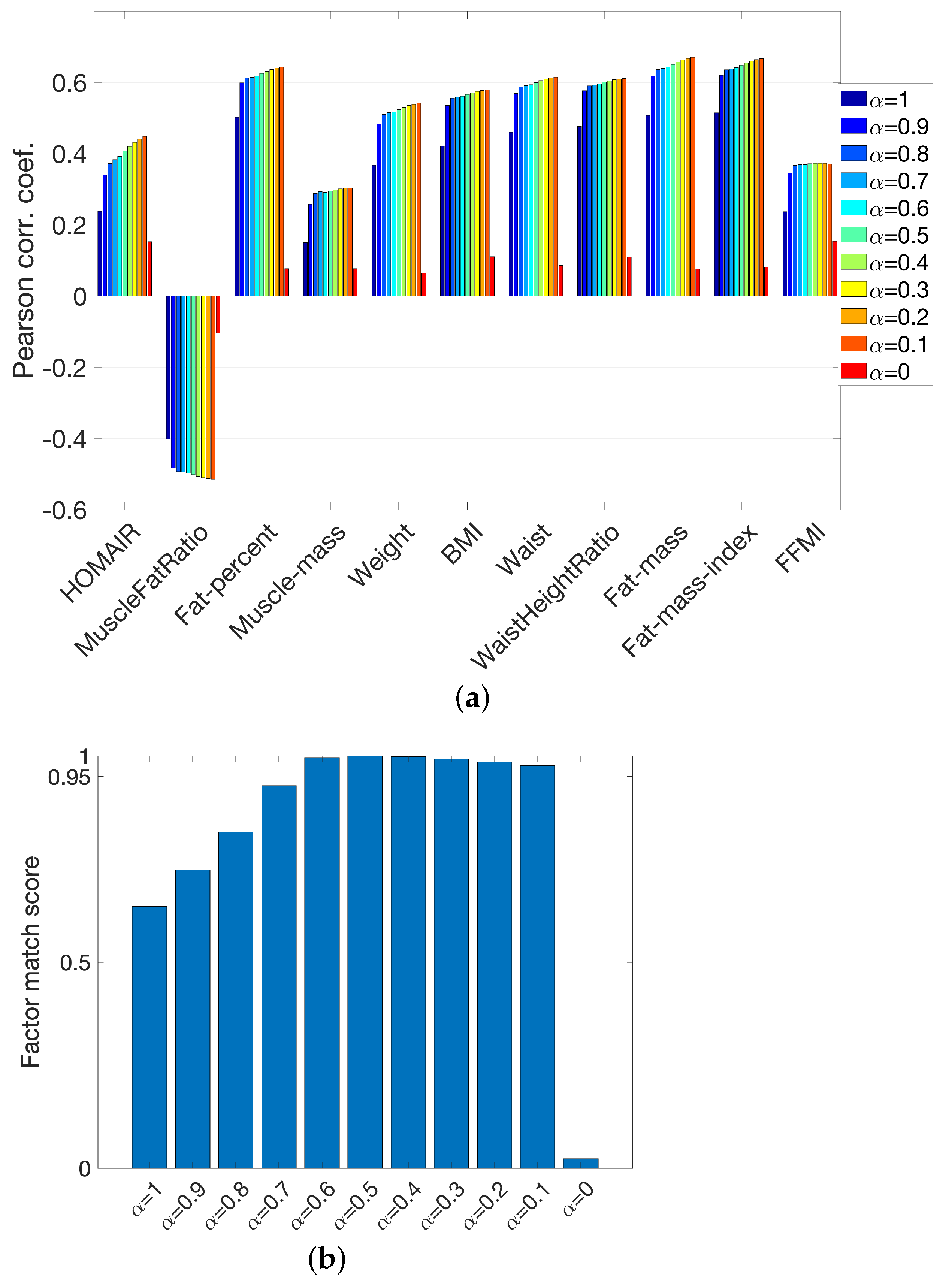
3.5. Joint Analysis of Real and Simulated Metabolomics Data in the Presence of Conflicting Information
- Step 1. Default patterns. We used a three-component CP model to extract the underlying patterns from the simulated T0-corrected data (see Figure S6a in Supplementary File). The data approximated by the model were denoted by , and residuals by .
- Step 2. Conflicting pattern construction. The first and third components (from Step 1) were retained, while the second component was modified by introducing wrong prior information. In the default (correct) pattern, Ins and Glc were close to each other, having large positive values in the second component, while values of the remaining metabolites were close to zero. We broke down the positive association between Ins and Glc and set the loading values of Ins, Glc, Pyr, Lac, Ala, and Bhb to 1, −1, 0, 0, 0, and 0, respectively (the factor vector was then normalized, i.e., divided by its two-norm). See Figure S6b in Supplementary File for the modified pattern. This is wrong prior information for the real data, which consisted of healthy subjects, and no such relation between Ins and Glc was expected.
- Step 3. Construction of simulated data with conflicting information. Tensor was then constructed using the modified CP patterns. The simulated data with conflicting information, denoted by , were obtained by adding the residual term (obtained in Step 1) to , i.e.,
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COPSAC | Copenhagen Prospective Studies on Asthma in Childhood |
| BMI | Body mass index |
| WBM | whole-body model |
| NMR | Nuclear Magnetic Resonance |
| HOMA-IR | Homeostatic model assessment for Insulin Resistance |
| Ins | Insulin |
| Glc | Glucose |
| Pyr | Pyruvate |
| Lac | Lactate |
| Ala | Alanine |
| Bhb | -hydroxybutyrate |
| CP | CANDECOMP/PARAFAC |
| CMTF | Coupled Matrix and Tensor Factorizations |
| ACMTF | Advanced Coupled Matrix and Tensor Factorizations |
| FMS | Factor match score |
| PINNs | Physics-informed neural networks |
| KGML | Knowledge-guided machine learning |
References
- Price, N.D.; Magis, A.T.; Earls, J.C.; Glusman, G.; Levy, R.; Lausted, C.; McDonald, D.T.; Kusebauch, U.; Moss, C.L.; Zhou, Y.; et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. 2017, 35, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Panyard, D.J.; Yu, B.; Snyder, M.P. The metabolomics of human aging: Advances, challenges, and opportunities. Sci. Adv. 2022, 8, eadd6155. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef]
- Swainston, N.; Smallbone, K.; Hefzi, H.; Dobson, P.D.; Brewer, J.; Hanscho, M.; Zielinski, D.C.; Ang, K.S.; Gardiner, N.J.; Gutierrez, J.M.; et al. Recon 2.2: From reconstruction to model of human metabolism. Metabolomics 2016, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M.T. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 2020, 16, e8982. [Google Scholar] [CrossRef]
- Kurata, H. Virtual metabolic human dynamic model for pathological analysis and therapy design for diabetes. iScience 2021, 24, 102101. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.; Snyder, M. Multi-Omics Profiling for Health. Mol. Cell. Proteom. 2023, 22, 100561. [Google Scholar] [CrossRef] [PubMed]
- Lépine, G.; Tremblay-Franco, M.; Bouder, S.; Dimina, L.; Fouillet, H.; Mariotti, F.; Polakof, S. Investigating the Postprandial Metabolome after Challenge Tests to Assess Metabolic Flexibility and Dysregulations Associated with Cardiometabolic Diseases. Nutrients 2022, 14, 472. [Google Scholar] [CrossRef]
- Yan, S.; Li, L.; Horner, D.; Ebrahimi, P.; Chawes, B.; Dragsted, L.O.; Rasmussen, M.A.; Smilde, A.K.; Acar, E. Characterizing human postprandial metabolic response using multiway data analysis. Metabolomics 2024, 20, 50. [Google Scholar] [CrossRef]
- Rozendaal, Y.J.W.; Wang, Y.; Paalvast, Y.; Tambyrajah, L.L.; Li, Z.; Willems van Dijk, K.; Rensen, P.C.N.; Kuivenhoven, J.A.; Groen, A.K.; Hilbers, P.A.J.; et al. In vivo and in silico dynamics of the development of Metabolic Syndrome. PLOS Comput. Biol. 2018, 14, e1006145. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, S.; Stroeve, J.H.M.; Stafleu, A.; Bakker, G.C.M.; Burggraaf, J.; van Erk, M.J.; Pellis, L.; Boessen, R.; Kardinaal, A.A.F.; van Ommen, B. Multi-parameter comparison of a standardized mixed meal tolerance test in healthy and type 2 diabetic subjects: The PhenFlex challenge. Genes Nutr. 2017, 12, 21. [Google Scholar] [CrossRef]
- Berry, S.E.; Valdes, A.M.; Drew, D.A.; Asnicar, F.; Mazidi, M.; Wolf, J.; Capdevila, J.; Hadjigeorgiou, G.; Davies, R.; Al Khatib, H.; et al. Human postprandial responses to food and potential for precision nutrition. Nat. Med. 2020, 26, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Pellis, L.; van Erk, M.J.; van Ommen, B.; Bakker, G.C.M.; Hendriks, H.F.J.; Cnubben, N.H.P.; Kleemann, R.; van Someren, E.P.; Bobeldijk, I.; Rubingh, C.M.; et al. Plasma metabolomics and proteomics profiling after a postprandial challenge reveal subtle diet effects on human metabolic status. Metabolomics 2012, 8, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, K.M.; Mazidi, M.; Franks, P.W.; Maher, T.; Valdes, A.M.; Linenberg, I.; Wolf, J.; Hadjigeorgiou, G.; Spector, T.D.; Menni, C.; et al. Characterisation of Fasting and Postprandial NMR Metabolites: Insights from the ZOE PREDICT 1 Study. Nutrients 2023, 15, 2638. [Google Scholar] [CrossRef] [PubMed]
- Blaise, B.J.; Correia, G.D.S.; Haggart, G.A.; Surowiec, I.; Sands, C.; Lewis, M.R.; Pearce, J.T.M.; Trygg, J.; Nicholson, J.K.; Holmes, E.; et al. Statistical analysis in metabolic phenotyping. Nat. Protoc. 2021, 16, 4299–4326. [Google Scholar] [CrossRef]
- Wojczynski, M.K.; Glasser, S.P.; Oberman, A.; Kabagambe, E.K.; Hopkins, P.N.; Tsai, M.Y.; Straka, R.J.; Ordovas, J.M.; Arnett, D.K. High-fat meal effect on LDL, HDL, and VLDL particle size and number in the Genetics of Lipid-Lowering Drugs and Diet Network (GOLDN): An interventional study. Lipids Health Dis. 2011, 10, 181. [Google Scholar] [CrossRef] [PubMed]
- Müllner, E.; Röhnisch, H.E.; Brömssen, C.V.; Moazzami, A.A. Metabolomics analysis reveals altered metabolites in lean compared with obese adolescents and additional metabolic shifts associated with hyperinsulinaemia and insulin resistance in obese adolescents: A cross-sectional study. Metabolomics 2021, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hoefsloot, H.; Graaf, A.A.; Acar, E.; Smilde, A.K. Exploring Dynamic Metabolomics Data With Multiway Data Analysis: A Simulation Study. BMC Bioinform. 2022, 23, 31. [Google Scholar] [CrossRef]
- Fujita, S.; Karasawa, Y.; Hironaka, K.; Taguchi, Y.; Kuroda, S. Features extracted using tensor decomposition reflect the biological features of the temporal patterns of human blood multimodal metabolome. PLoS ONE 2023, 18, e0281594. [Google Scholar] [CrossRef] [PubMed]
- Skantze, V.; Wallman, M.; Sandberg, A.S.; Landberg, R.; Jirstrand, M.; Brunius, C. Identification of metabotypes in complex biological data using tensor decomposition. Chemom. Intell. Lab. Syst. 2023, 233, 104733. [Google Scholar] [CrossRef]
- von Rueden, L.; Mayer, S.; Beckh, K.; Georgiev, B.; Giesselbach, S.; Heese, R.; Kirsch, B.; Pfrommer, J.; Pick, A.; Ramamurthy, R.; et al. Informed Machine Learning – A Taxonomy and Survey of Integrating Prior Knowledge into Learning Systems. IEEE Trans. Knowl. Data Eng. 2023, 35, 614–633. [Google Scholar] [CrossRef]
- Raissi, M.; Perdikaris, P.; Karniadakis, G. Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. J. Comput. Phys. 2019, 378, 686–707. [Google Scholar] [CrossRef]
- Karpatne, A.; Jia, X.; Kumar, V. Knowledge-guided Machine Learning: Current Trends and Future Prospects. arXiv 2024, arXiv:2403.15989v2. [Google Scholar]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2019, 48, D445–D453. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2022, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H. The Copenhagen Prospective Study on Asthma in Childhood (COPSAC): Design, rationale, and baseline data from a longitudinal birth cohort study. Ann. Allergy Asthma Immunol. 2004, 93, 381–389. [Google Scholar] [CrossRef]
- Stroeve, J.H.M.; Wietmarschen, H.V.; Kremer, B.H.A.; Ommen, B.V.; Wopereis, S. Phenotypic flexibility as a measure of health: The optimal nutritional stress response test. Genes Nutr. 2015, 10, 1–21. [Google Scholar] [CrossRef]
- Li, L.; Yan, S.; Horner, D.; Rasmussen, M.A.; Smilde, A.K.; Acar, E. Revealing static and dynamic biomarkers from postprandial metabolomics data through coupled matrix and tensor factorizations. Metabolomics 2024, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yan, S.; Bakker, B.M.; Hoefsloot, H.; Chawes, B.; Horner, D.; Rasmussen, M.A.; Smilde, A.K.; Acar, E. Analyzing postprandial metabolomics data using multiway models: A simulation study. BMC Bioinform. 2024, 25. [Google Scholar] [CrossRef]
- Acar, E.; Yener, B. Unsupervised Multiway Data Analysis: A Literature Survey. IEEE Trans. Knowl. Data Eng. 2009, 21, 6–20. [Google Scholar] [CrossRef]
- Kolda, T.G.; Bader, B.W. Tensor Decompositions and Applications. SIAM Rev. 2009, 51, 455–500. [Google Scholar] [CrossRef]
- Acar, E.; Bingol, C.A.; Bingol, H.; Bro, R.; Yener, B. Multiway Analysis of Epilepsy Tensors. Bioinformatics 2007, 23, i10–i18. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Kim, T.H.; Wang, F.; Vyas, S.; Ryu, S.I.; Shenoy, K.V.; Schnitzer, M.; Kolda, T.G.; Ganguli, S. Unsupervised Discovery of Demixed, Low-Dimensional Neural Dynamics across Multiple Timescales through Tensor Component Analysis. Neuron 2018, 98, 1099–1115.e8. [Google Scholar] [CrossRef] [PubMed]
- Smilde, A.K.; Geladi, P.; Bro, R. Multi-Way Analysis with Applications in the Chemical Sciences; Wiley: West Sussex, UK, 2004. [Google Scholar]
- Papalexakis, E.E.; Faloutsos, C.; Sidiropoulos, N.D. Tensors for Data Mining and Data Fusion: Models, Applications, and Scalable Algorithms. ACM Trans. Intell. Syst. Technol. 2016, 8, 1–44. [Google Scholar] [CrossRef]
- Carroll, J.D.; Chang, J.J. Analysis of individual differences in multidimensional scaling via an N-way generalization of “Eckart-Young” decomposition. Psychometrika 1970, 35, 283–319. [Google Scholar] [CrossRef]
- Harshman, R.A. Foundations of the PARAFAC procedure: Models and conditions for an “explanatory” multi-modal factor analysis. UCLA Work. Pap. Phon. 1970, 16, 84. [Google Scholar]
- Hitchcock, F.L. The Expression of a Tensor or a Polyadic as a Sum of Products. J. Math. Phys. 1927, 6, 164–189. [Google Scholar] [CrossRef]
- Acar, E.; Dunlavy, D.M.; Kolda, T.G.; Mørup, M. Scalable tensor factorizations for incomplete data. Chemom. Intell. Lab. Syst. 2011, 106, 41–56. [Google Scholar] [CrossRef]
- Martino, C.; Shenhav, L.; Marotz, C.; Armstrong, G.; McDonald, D.; Vázquez-Baeza, Y.; Morton, J.T.; Jiang, L.; Dominguez-Bello, M.G.; Swafford, A.D.; et al. Context-aware dimensionality reduction deconvolutes gut microbial community dynamics. Nat. Biotechnol. 2021, 39, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Hunyadi, B.; Dupont, P.; Van Paesschen, W.; Van Huffel, S. Tensor decompositions and data fusion in epileptic electroencephalography and functional magnetic resonance imaging data. WIREs Data Min. Knowl. Discov. 2017, 7, e1197. [Google Scholar] [CrossRef]
- Acar, E.; Bro, R.; Smilde, A.K. Data Fusion in Metabolomics using Coupled Matrix and Tensor Factorizations. Proc. IEEE 2015, 103, 1602–1620. [Google Scholar] [CrossRef]
- Acar, E.; Kolda, T.G.; Dunlavy, D.M. All-at-once Optimization For Coupled Matrix and Tensor Factorizations. arXiv 2011, arXiv:1105.3422. [Google Scholar]
- Sørensen, M.; Lathauwer, L.D.D. Coupled Canonical Polyadic Decompositions and (Coupled) Decompositions in Multilinear Rank-(Lr,n,Lr,n,1) Terms—Part I: Uniqueness. SIAM J. Matrix Anal. Appl. 2015, 36, 496–522. [Google Scholar] [CrossRef]
- Acar, E.; Papalexakis, E.E.; Gurdeniz, G.; Rasmussen, M.A.; Lawaetz, A.J.; Nilsson, M.; Bro, R. Structure-Revealing Data Fusion. BMC Bioinform. 2014, 15, 239. [Google Scholar] [CrossRef] [PubMed]
- Kanatsoulis, C.I.; Fu, X.; Sidiropoulos, N.D.; Ma, W.K. Hyperspectral Super-Resolution: A Coupled Tensor Factorization Approach. IEEE Trans. Signal Process. 2018, 66, 6503–6517. [Google Scholar] [CrossRef]
- Acar, E.; Schenker, C.; Levin-Schwartz, Y.; Calhoun, V.; Adali, T. Unraveling Diagnostic Biomarkers of Schizophrenia through Structure-Revealing Fusion of Multi-Modal Neuroimaging Data. Front. Neurosci. 2019, 13, 416. [Google Scholar] [CrossRef]
- Ermis, B.; Acar, E.; Cemgil, A.T. Link prediction in heterogeneous data via generalized coupled tensor factorization. Data Min. Knowl. Discov. 2015, 29, 203–236. [Google Scholar] [CrossRef]
- Afshar, A.; Perros, I.; Park, H.; Defilippi, C.; Yan, X.; Stewart, W.; Ho, J.; Sun, J. Taste: Temporal and static tensor factorization for phenotyping electronic health records. In Proceedings of the ACM Conference on Health, Inference, and Learning, Toronto, ON, Canada, 2–4 April 2020; pp. 193–203. [Google Scholar]
- Bro, R.; Smilde, A.K. Centering and scaling in component analysis. J. Chemom. 2003, 17, 16–33. [Google Scholar] [CrossRef]
- Bader, B.W.; Kolda, T.G. Matlab Tensor Toolbox, Version 3.1. Available online: https://www.tensortoolbox.org (accessed on 19 December 2024).
- Dunlavy, D.M.; Kolda, T.G.; Acar, E. Poblano v1.0: A Matlab Toolbox for Gradient-Based Optimization; Technical Repor; Sandia National Laboratories: Albuquerque, NM, USA, 2010. [Google Scholar]
- Adali, T.; Kantar, F.; Akhonda, M.A.B.S.; Strother, S.; Calhoun, V.D.; Acar, E. Reproducibility in Matrix and Tensor Decompositions: Focus on model match, interpretability, and uniqueness. IEEE Signal Process. Mag. 2022, 39, 8–24. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Li-Gao, R.; Bull, C.J.; de Mutsert, R.; Rosendaal, F.R.; Mook-Kanamori, D.O.; Willems van Dijk, K.; Timpson, N.J. The association between body mass index and metabolite response to a liquid mixed meal challenge: A Mendelian randomization study. Am. J. Clin. Nutr. 2024, 119, 1354–1370. [Google Scholar] [CrossRef]
- Wilderjans, T.F.; Ceulemans, E.; Mechelen, I.V.; van den Berg, R.A. Simultaneous analysis of coupled data matrices subject to different amounts of noise. Br. J. Math. Stat. Psychol. 2011, 64, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Simsekli, U.; Ermis, B.; Cemgil, A.T.; Acar, E. Optimal Weight Learning for Coupled Tensor Factorization with Mixed Divergences. In Proceedings of the EUSIPCO’13: Proceedings of 21st European Signal Processing Conference, Marrakech, Morocco, 9–13 September 2013; pp. 1–5. [Google Scholar]
- Schenker, C.; Cohen, J.E.; Acar, E. A Flexible Optimization Framework for Regularized Matrix-Tensor Factorizations with Linear Couplings. IEEE J. Sel. Top. Signal Process. 2021, 15, 506–521. [Google Scholar] [CrossRef]
- Khan, S.A.; Leppaaho, E.; Kaski, S. Bayesian multi-tensor factorization. Mach. Learn. 2016, 105, 233–253. [Google Scholar] [CrossRef]
- Babbar, V.; Guo, Z.; Rudin, C. What is different between these datasets? arXiv 2024, arXiv:2403.05652. [Google Scholar]
- Shen, X.; Kellogg, R.; Panyard, D.J.; Bararpour, N.; Castillo, K.E.; Lee-McMullen, B.; Delfarah, A.; Ubellacker, J.; Ahadi, S.; Rosenberg-Hasson, Y.; et al. Multi-omics microsampling for the profiling of lifestyle-associated changes in health. Nat. Biomed. Eng. 2023, 8, 11–29. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Hoefsloot, H.; Bakker, B.M.; Horner, D.; Rasmussen, M.A.; Smilde, A.K.; Acar, E. Longitudinal Metabolomics Data Analysis Informed by Mechanistic Models. Metabolites 2025, 15, 2. https://doi.org/10.3390/metabo15010002
Li L, Hoefsloot H, Bakker BM, Horner D, Rasmussen MA, Smilde AK, Acar E. Longitudinal Metabolomics Data Analysis Informed by Mechanistic Models. Metabolites. 2025; 15(1):2. https://doi.org/10.3390/metabo15010002
Chicago/Turabian StyleLi, Lu, Huub Hoefsloot, Barbara M. Bakker, David Horner, Morten A. Rasmussen, Age K. Smilde, and Evrim Acar. 2025. "Longitudinal Metabolomics Data Analysis Informed by Mechanistic Models" Metabolites 15, no. 1: 2. https://doi.org/10.3390/metabo15010002
APA StyleLi, L., Hoefsloot, H., Bakker, B. M., Horner, D., Rasmussen, M. A., Smilde, A. K., & Acar, E. (2025). Longitudinal Metabolomics Data Analysis Informed by Mechanistic Models. Metabolites, 15(1), 2. https://doi.org/10.3390/metabo15010002