
The Role of Lysophospholipid Metabolites LPC and LPA in the Pathogenesis of Chronic Obstructive Pulmonary Disease


Abstract
1. Introduction
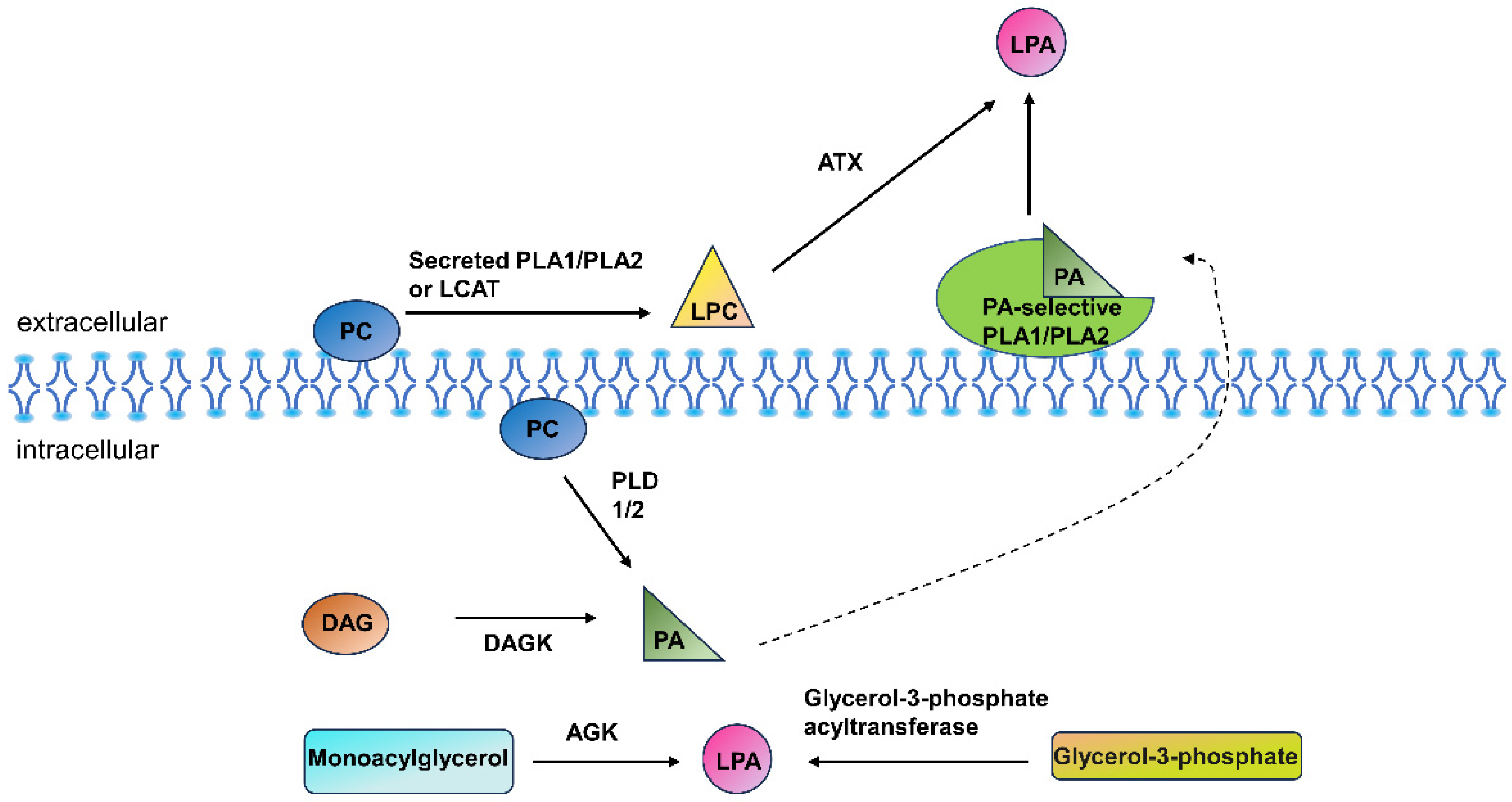
2. Lysophospholipids
3. LPA, LPC Receptors in the Respiratory System
3.1. LPA
3.2. LPC
4. Role of LPC–LPA Axis in COPD Pathogenesis
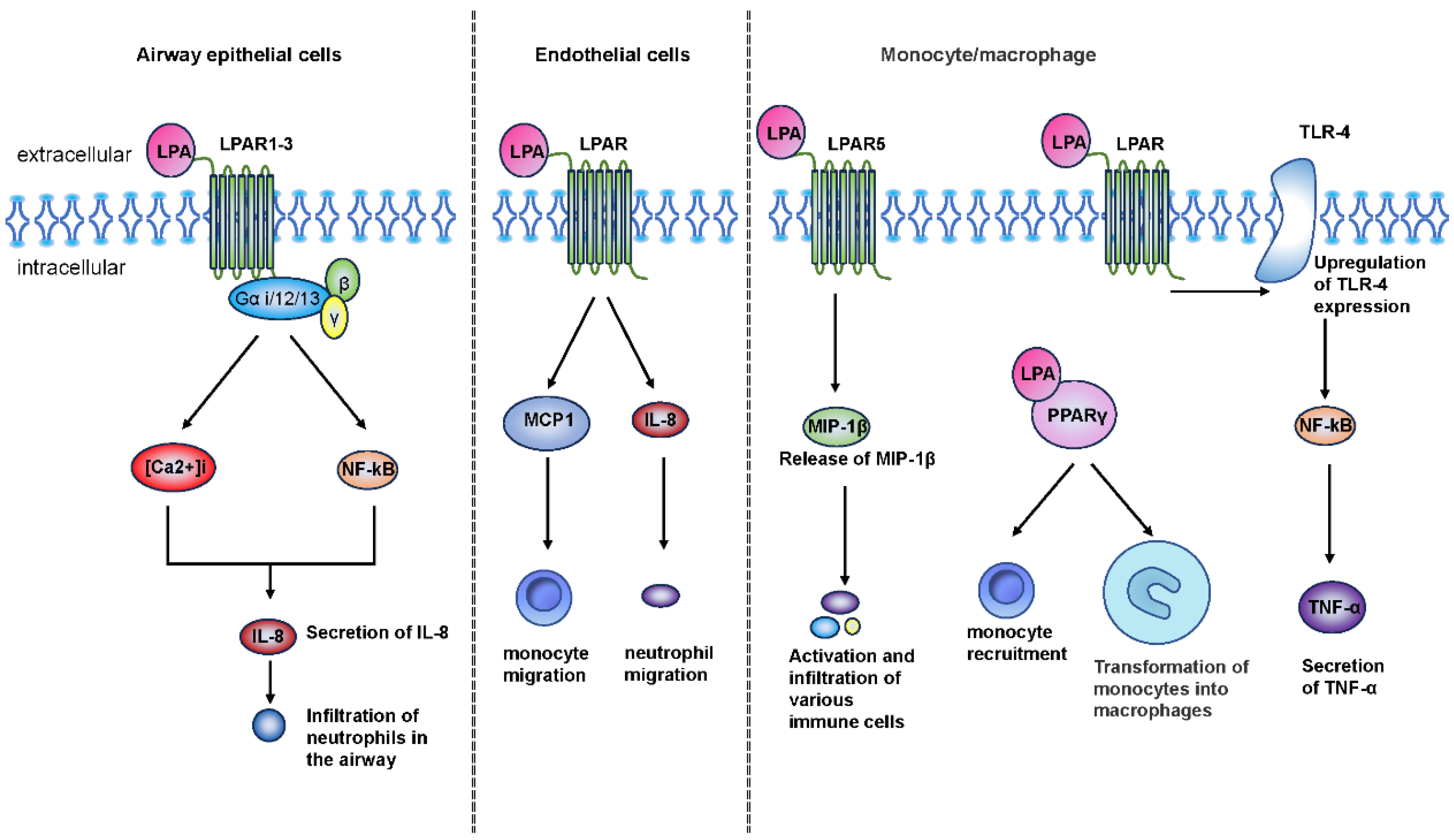
4.1. Chronic Airway Inflammation
4.2. Airway Remodeling and Fibrosis
4.3. Apoptosis
4.4. Oxidative Stress
5. Association of Lysophospholipids with Clinical Features, Phenotypes, and Prognosis of COPD
6. Potential Therapeutic Targets
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease—2024 Report. 2024. Available online: https://goldcopd.org/2024-gold-report/ (accessed on 2 March 2024).
- Wang, C.; Xu, J.; Yang, L.; Xu, Y.; Zhang, X.; Bai, C.; Kang, J.; Ran, P.; Shen, H.; Wen, F.; et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): A national cross-sectional study. Lancet 2018, 391, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Guo, C.; Zhang, L.; Zhang, L.; Abulikemu, M.; Wang, J.; Zhou, Q.; Chen, Y.; Sun, Y.; Chang, C. Serum Glycerophospholipid Profile in Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Front. Physiol. 2021, 12, 646010. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Pathogenesis of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 258–266; discussion 290–291. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Pavlisko, E.; Voynow, J.A. Pathogenic triad in COPD: Oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chron. Obstr. Pulm. Dis. 2011, 6, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, Z.; Dong, L.; Wu, Y.; Shen, H.; Chen, Z. Lipid metabolism in chronic obstructive pulmonary disease. Int. J. Chron. Obstr. Pulm. Dis. 2019, 14, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.T.; Ramesh, T.; Toh, X.R.; Nguyen, L.N. Emerging roles of lysophospholipids in health and disease. Progress. Lipid Res. 2020, 80, 101068. [Google Scholar] [CrossRef] [PubMed]
- Sevastou, I.; Kaffe, E.; Mouratis, M.A.; Aidinis, V. Lysoglycerophospholipids in chronic inflammatory disorders: The PLA(2)/LPC and ATX/LPA axes. Biochim. Biophys. Acta 2013, 1831, 42–60. [Google Scholar] [CrossRef]
- Rindlisbacher, B.; Schmid, C.; Geiser, T.; Bovet, C.; Funke-Chambour, M. Serum metabolic profiling identified a distinct metabolic signature in patients with idiopathic pulmonary fibrosis—A potential biomarker role for LysoPC. Respir. Res. 2018, 19, 7. [Google Scholar] [CrossRef]
- Kano, K.; Aoki, J.; Hla, T. Lysophospholipid Mediators in Health and Disease. Annu. Rev. Pathol. 2022, 17, 459–483. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, S.J.; Park, G.Y.; Christman, J.W.; Nyenhuis, S.; Berdyshev, E.; Natarajan, V. Polyunsaturated lysophosphatidic acid as a potential asthma biomarker. Biomark. Med. 2016, 10, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Payne, S.G.; Liu, H.; Goparaju, S.; Milstien, S.; Spiegel, S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J. Cell Biol. 2005, 169, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Hosogaya, S.; Yatomi, Y.; Nakamura, K.; Ohkawa, R.; Okubo, S.; Yokota, H.; Ohta, M.; Yamazaki, H.; Koike, T.; Ozaki, Y. Measurement of plasma lysophosphatidic acid concentration in healthy subjects: Strong correlation with lysophospholipase D activity. Ann. Clin. Biochem. 2008, 45 Pt 4, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, K.; Valentine, W.J. Druggable Lysophospholipid Signaling Pathways. Adv. Exp. Med. Biol. 2020, 1274, 137–176. [Google Scholar] [PubMed]
- Shea, B.S.; Tager, A.M. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hla, T.; Lynch, K.R.; Spiegel, S.; Moolenaar, W.H. International union of basic and clinical pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2010, 62, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.A.; Fredrickson, M.A.; D’Antonio, M.; Katsnelson, E.; MacBeth, M.; Van Gulick, R.; Chimed, T.S.; McCarter, M.; D’Alessandro, A.; Robinson, W.A.; et al. Lysophosphatidic acid modulates CD8 T cell immunosurveillance and metabolism to impair anti-tumor immunity. Nat. Commun. 2023, 14, 3214. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.F.; Nanayakkara, G.; Shao, Y.; Liang, B.; Cole, L.; Yang, W.Y.; Li, X.; Cueto, R.; Yu, J.; et al. Lysophospholipid Receptors, as Novel Conditional Danger Receptors and Homeostatic Receptors Modulate Inflammation-Novel Paradigm and Therapeutic Potential. J. Cardiovasc. Transl. Res. 2016, 9, 343–359. [Google Scholar] [CrossRef]
- Anliker, B.; Chun, J. Lysophospholipid G protein-coupled receptors. J. Biol. Chem. 2004, 279, 20555–20558. [Google Scholar] [CrossRef]
- Meduri, B.; Pujar, G.V.; Durai Ananda Kumar, T.; Akshatha, H.S.; Sethu, A.K.; Singh, M.; Kanagarla, A.; Mathew, B. Lysophosphatidic acid (LPA) receptor modulators: Structural features and recent development. Eur. J. Med. Chem. 2021, 222, 113574. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Natarajan, V. Lysophosphatidic acid (LPA) and its receptors: Role in airway inflammation and remodeling. Biochim. Biophys. Acta 2013, 1831, 86–92. [Google Scholar] [CrossRef]
- Sheng, X.; Yung, Y.C.; Chen, A.; Chun, J. Lysophosphatidic acid signalling in development. Development 2015, 142, 1390–1395. [Google Scholar] [CrossRef]
- Cummings, R.; Zhao, Y.; Jacoby, D.; Spannhake, E.W.; Ohba, M.; Garcia, J.G.; Watkins, T.; He, D.; Saatian, B.; Natarajan, V. Protein kinase Cdelta mediates lysophosphatidic acid-induced NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. J. Biol. Chem. 2004, 279, 41085–41094. [Google Scholar] [CrossRef]
- Blaho, V.A.; Chun, J. ‘Crystal’ Clear? Lysophospholipid Receptor Structure Insights and Controversies. Trends Pharmacol. Sci. 2018, 39, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, D.; Zhao, J.; Wang, L.; Leff, A.R.; Spannhake, E.W.; Georas, S.; Natarajan, V. Lysophosphatidic acid induces interleukin-13 (IL-13) receptor alpha2 expression and inhibits IL-13 signaling in primary human bronchial epithelial cells. J. Biol. Chem. 2007, 282, 10172–10179. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Knudsen, E.; Jin, Y.; Gessani, S.; Maghazachi, A.A. Lysophospholipids and chemokines activate distinct signal transduction pathways in T helper 1 and T helper 2 cells. Cell. Signal. 2004, 16, 991–1000. [Google Scholar] [CrossRef]
- Kotarsky, K.; Boketoft, A.; Bristulf, J.; Nilsson, N.E.; Norberg, A.; Hansson, S.; Owman, C.; Sillard, R.; Leeb-Lundberg, L.M.; Olde, B. Lysophosphatidic acid binds to and activates GPR92, a G protein-coupled receptor highly expressed in gastrointestinal lymphocytes. J. Pharmacol. Exp. Ther. 2006, 318, 619–628. [Google Scholar] [CrossRef]
- Park, G.Y.; Lee, Y.G.; Berdyshev, E.; Nyenhuis, S.; Du, J.; Fu, P.; Gorshkova, I.A.; Li, Y.; Chung, S.; Karpurapu, M.; et al. Autotaxin production of lysophosphatidic acid mediates allergic asthmatic inflammation. Am. J. Respir. Crit. Care Med. 2013, 188, 928–940. [Google Scholar] [CrossRef]
- Liu, P.; Zhu, W.; Chen, C.; Yan, B.; Zhu, L.; Chen, X.; Peng, C. The mechanisms of lysophosphatidylcholine in the development of diseases. Life Sci. 2020, 247, 117443. [Google Scholar] [CrossRef]
- Radu, C.G.; Yang, L.V.; Riedinger, M.; Au, M.; Witte, O.N. T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.V.; Radu, C.G.; Wang, L.; Riedinger, M.; Witte, O.N. Gi-independent macrophage chemotaxis to lysophosphatidylcholine via the immunoregulatory GPCR G2A. Blood 2005, 105, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.B.; Iaciura, B.M.; Nohara, L.L.; Lopes, C.D.; Veas, E.M.; Mariano, V.S.; Bozza, P.T.; Lopes, U.G.; Atella, G.C.; Almeida, I.C.; et al. Lysophosphatidylcholine triggers TLR2- and TLR4-mediated signaling pathways but counteracts LPS-induced NO synthesis in peritoneal macrophages by inhibiting NF-κB translocation and MAPK/ERK phosphorylation. PLoS ONE 2013, 8, e76233. [Google Scholar] [CrossRef]
- Magalhães, K.; Almeida, P.E.; Atella, G.; Maya-Monteiro, C.M.; Castro-Faria-Neto, H.; Pelajo-Machado, M.; Lenzi, H.L.; Bozza, M.T.; Bozza, P.T. Schistosomal-derived lysophosphatidylcholine are involved in eosinophil activation and recruitment through Toll-like receptor-2-dependent mechanisms. J. Infect. Dis. 2010, 202, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Song, M.H.; Gupta, A.; Kim, H.O.; Oh, K. Lysophosphatidylcholine aggravates contact hypersensitivity by promoting neutrophil infiltration and IL17 expression. BMB Rep. 2021, 54, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. (Lond.) 2017, 131, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Natarajan, V. Lysophosphatidic acid signaling in airway epithelium: Role in airway inflammation and remodeling. Cell. Signal. 2009, 21, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Saatian, B.; Zhao, Y.; He, D.; Georas, S.N.; Watkins, T.; Spannhake, E.W.; Natarajan, V. Transcriptional regulation of lysophosphatidic acid-induced interleukin-8 expression and secretion by p38 MAPK and JNK in human bronchial epithelial cells. Biochem. J. 2006, 393 Pt 3, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, D.; Saatian, B.; Watkins, T.; Spannhake, E.W.; Pyne, N.J.; Natarajan, V. Regulation of lysophosphatidic acid-induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase Cdelta, Lyn kinase, and matrix metalloproteinases. J. Biol. Chem. 2006, 281, 19501–19511. [Google Scholar] [CrossRef]
- Zhao, Y.; Usatyuk, P.V.; Cummings, R.; Saatian, B.; He, D.; Watkins, T.; Morris, A.; Spannhake, E.W.; Brindley, D.N.; Natarajan, V. Lipid phosphate phosphatase-1 regulates lysophosphatidic acid-induced calcium release, NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. Biochem. J. 2005, 385 Pt 2, 493–502. [Google Scholar] [CrossRef]
- Rahaman, M.; Costello, R.W.; Belmonte, K.E.; Gendy, S.S.; Walsh, M.T. Neutrophil sphingosine 1-phosphate and lysophosphatidic acid receptors in pneumonia. Am. J. Respir. Cell Mol. Biol. 2006, 34, 233–241. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Su, Y.; Usatyuk, P.V.; Spannhake, E.W.; Kogut, P.; Solway, J.; Natarajan, V.; Zhao, Y. Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury. J. Biol. Chem. 2009, 284, 24123–24132. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Matoba, K.; Kawanami, D.; Nagai, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 1331. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Rai, V. Lysophosphatidic acid converts monocytes into macrophages in both mice and humans. Blood 2017, 129, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wong, W.; Birnberg, A.; Chakrabarti, A.; Yang, X.; Choy, D.F.; Olsson, J.; Verschueren, E.; Neighbors, M.; Sandoval, W.; et al. Lysophosphatidic acid species are associated with exacerbation in chronic obstructive pulmonary disease. BMC Pulm. Med. 2021, 21, 301. [Google Scholar] [CrossRef] [PubMed]
- Gustin, C.; Van Steenbrugge, M.; Raes, M. LPA modulates monocyte migration directly and via LPA-stimulated endothelial cells. Am. J. Physiol. Cell Physiol. 2008, 295, C905–C914. [Google Scholar] [CrossRef] [PubMed]
- Lundequist, A.; Boyce, J.A. LPA5 is abundantly expressed by human mast cells and important for lysophosphatidic acid induced MIP-1β release. PLoS ONE 2011, 6, e18192. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhou, Z.; Li, X.; Niu, J. The effect of lysophosphatidic acid on Toll-like receptor 4 expression and the nuclear factor-κB signaling pathway in THP-1 cells. Mol. Cell Biochem. 2016, 422, 41–49. [Google Scholar] [CrossRef]
- He, D.; Natarajan, V.; Stern, R.; Gorshkova, I.A.; Solway, J.; Spannhake, E.W.; Zhao, Y. Lysophosphatidic acid-induced transactivation of epidermal growth factor receptor regulates cyclo-oxygenase-2 expression and prostaglandin E(2) release via C/EBPbeta in human bronchial epithelial cells. Biochem. J. 2008, 412, 153–162. [Google Scholar] [CrossRef]
- Vancheri, C.; Mastruzzo, C.; Sortino, M.A.; Crimi, N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004, 25, 40–46. [Google Scholar] [CrossRef]
- Nakata, J.; Kondo, M.; Tamaoki, J.; Takemiya, T.; Nohara, M.; Yamagata, K.; Nagai, A. Augmentation of allergic inflammation in the airways of cyclooxygenase-2-deficient mice. Respirology 2005, 10, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, G.; Sandhya Rani, M.R.; Gerber, C.E.; Mukhopadhyay, C.; Ransohoff, R.M.; Chisolm, G.M.; Kottke-Marchant, K. Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. J. Mol. Cell. Cardiol. 2003, 35, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Kusumi, Y.; Mitsumata, M.; Li, X.K.; Fujino, M. Lysophosphatidylcholine upregulates LOX-1, chemokine receptors, and activation-related transcription factors in human T-cell line Jurkat. J. Thromb. Thrombolysis 2008, 26, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Rolin, J.; Vego, H.; Maghazachi, A.A. Oxidized lipids and lysophosphatidylcholine induce the chemotaxis, up-regulate the expression of CCR9 and CXCR4 and abrogate the release of IL-6 in human monocytes. Toxins 2014, 6, 2840–2856. [Google Scholar] [CrossRef]
- Tan, M.; Hao, F.; Xu, X.; Chisolm, G.M.; Cui, M.Z. Lysophosphatidylcholine activates a novel PKD2-mediated signaling pathway that controls monocyte migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Eder, C. Lysophosphatidylcholine- and MCP-1-induced chemotaxis of monocytes requires potassium channel activity. Pflug. Arch. 2009, 459, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Oestvang, J.; Anthonsen, M.W.; Johansen, B. LysoPC and PAF Trigger Arachidonic Acid Release by Divergent Signaling Mechanisms in Monocytes. J. Lipids 2011, 2011, 532145. [Google Scholar] [CrossRef] [PubMed]
- Bach, G.; Perrin-Cocon, L.; Gerossier, E.; Guironnet-Paquet, A.; Lotteau, V.; Inchauspé, G.; Fournillier, A. Single lysophosphatidylcholine components exhibit adjuvant activities in vitro and in vivo. Clin. Vaccine Immunol. 2010, 17, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef]
- Nan, W.; Xiong, F.; Zheng, H.; Li, C.; Lou, C.; Lei, X.; Wu, H.; Gao, H.; Li, Y. Myristoyl lysophosphatidylcholine is a biomarker and potential therapeutic target for community-acquired pneumonia. Redox Biol. 2022, 58, 102556. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Fu, L.; Cao, W.; Hu, B.; Zhao, H.; Li, J.B. Low Vitamin D Status Is Associated with Epithelial-Mesenchymal Transition in Patients with Chronic Obstructive Pulmonary Disease. J. Immunol. 2019, 203, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Machen, M.; Lange, M.; Exley, M.; Wu, S.; Usheva, A.; Robson, S.C. Lysophosphatidic acid generation by pulmonary NKT cell ENPP-2/autotaxin exacerbates hyperoxic lung injury. Purinergic Signal 2015, 11, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Ediger, T.L.; Schulte, N.A.; Murphy, T.J.; Toews, M.L. Transcription factor activation and mitogenic synergism in airway smooth muscle cells. Eur. Respir. J. 2003, 21, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Ediger, T.L.; Toews, M.L. Synergistic stimulation of airway smooth muscle cell mitogenesis. J. Pharmacol. Exp. Ther. 2000, 294, 1076–1082. [Google Scholar] [PubMed]
- Hirshman, C.A.; Emala, C.W. Actin reorganization in airway smooth muscle cells involves Gq and Gi-2 activation of Rho. Am. J. Physiol. 1999, 277, L653–L661. [Google Scholar] [PubMed]
- Toews, M.L.; Ediger, T.L.; Romberger, D.J.; Rennard, S.I. Lysophosphatidic acid in airway function and disease. Biochim. Biophys. Acta 2002, 1582, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, D.; Berdyshev, E.; Zhong, M.; Salgia, R.; Morris, A.J.; Smyth, S.S.; Natarajan, V.; Zhao, Y. Autotaxin induces lung epithelial cell migration through lysoPLD activity-dependent and -independent pathways. Biochem. J. 2011, 439, 45–55. [Google Scholar] [CrossRef]
- Decato, B.E.; Leeming, D.J.; Sand, J.M.B.; Fischer, A.; Du, S.; Palmer, S.M.; Karsdal, M.; Luo, Y.; Minnich, A. LPA(1) antagonist BMS-986020 changes collagen dynamics and exerts antifibrotic effects in vitro and in patients with idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 61. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Holmes, M.; Reynolds, P.N. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur. Respir. J. 2005, 25, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Petrache, I.; Elias, J.A.; Voelkel, N.F.; Henson, P.M. Apoptosis and emphysema: The missing link. Am. J. Respir. Cell Mol. Biol. 2003, 28, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, W.H.; Kruijer, W.; Tilly, B.C.; Verlaan, I.; Bierman, A.J.; de Laat, S.W. Growth factor-like action of phosphatidic acid. Nature 1986, 323, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Ediger, T.L.; Toews, M.L. Dual effects of lysophosphatidic acid on human airway smooth muscle cell proliferation and survival. Biochim. Biophys. Acta 2001, 1531, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Fu, P.; Patel, P.; Harijith, A.; Sun, T.; Zhao, Y.; Garcia, J.G.; Chun, J.; Natarajan, V. Lysophosphatidic acid receptor-2 deficiency confers protection against bleomycin-induced lung injury and fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Scicchitano, R.; Reynolds, P.N.; Holmes, M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell Biol. 2003, 81, 289–296. [Google Scholar] [CrossRef]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Barnawi, J.; Tran, H.B.; Roscioli, E.; Hodge, G.; Jersmann, H.; Haberberger, R.; Hodge, S. Pro-phagocytic Effects of Thymoquinone on Cigarette Smoke-exposed Macrophages Occur by Modulation of the Sphingosine-1-phosphate Signalling System. COPD 2016, 13, 653–661. [Google Scholar] [CrossRef]
- Morimoto, K.; Janssen, W.J.; Fessler, M.B.; McPhillips, K.A.; Borges, V.M.; Bowler, R.P.; Xiao, Y.Q.; Kench, J.A.; Henson, P.M.; Vandivier, R.W. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J. Immunol. 2006, 176, 7657–7665. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.A.; Kim, J.A.; Park, M.H.; Jung, S.C.; Suh, S.H.; Pang, M.G.; Kim, Y.J. Lysophosphatidylcholine induces endothelial cell injury by nitric oxide production through oxidative stress. J. Matern. Fetal Neonatal Med. 2009, 22, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, G.R. TRPC1/TRPC3 channels mediate lysophosphatidylcholine-induced apoptosis in cultured human coronary artery smooth muscles cells. Oncotarget 2016, 7, 50937–50951. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, J.; Ma, B.; Wang, J.; Chen, J. Role of Autophagy in Lysophosphatidylcholine-Induced Apoptosis of Mouse Ovarian Granulosa Cells. Int. J. Mol. Sci. 2022, 23, 1479. [Google Scholar] [CrossRef] [PubMed]
- Tanosaki, T.; Mikami, Y.; Shindou, H.; Suzuki, T.; Hashidate-Yoshida, T.; Hosoki, K.; Kagawa, S.; Miyata, J.; Kabata, H.; Masaki, K.; et al. Lysophosphatidylcholine Acyltransferase 1 Deficiency Promotes Pulmonary Emphysema via Apoptosis of Alveolar Epithelial Cells. Inflammation 2022, 45, 1765–1779. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Nanayakkara, G.; Cheng, J.; Cueto, R.; Yang, W.Y.; Park, J.Y.; Wang, H.; Yang, X. Lysophospholipids and Their Receptors Serve as Conditional DAMPs and DAMP Receptors in Tissue Oxidative and Inflammatory Injury. Antioxid. Redox Signal 2018, 28, 973–986. [Google Scholar] [CrossRef]
- Inoue, N.; Takeshita, S.; Gao, D.; Ishida, T.; Kawashima, S.; Akita, H.; Tawa, R.; Sakurai, H.; Yokoyama, M. Lysophosphatidylcholine increases the secretion of matrix metalloproteinase 2 through the activation of NADH/NADPH oxidase in cultured aortic endothelial cells. Atherosclerosis 2001, 155, 45–52. [Google Scholar] [CrossRef]
- Ares, M.P.; Kallin, B.; Eriksson, P.; Nilsson, J. Oxidized LDL induces transcription factor activator protein-1 but inhibits activation of nuclear factor-kappa B in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1584–1590. [Google Scholar] [CrossRef]
- Ojala, P.J.; Hirvonen, T.E.; Hermansson, M.; Somerharju, P.; Parkkinen, J. Acyl chain-dependent effect of lysophosphatidylcholine on human neutrophils. J. Leukoc. Biol. 2007, 82, 1501–1509. [Google Scholar] [CrossRef]
- Vázquez-Medina, J.P.; Dodia, C.; Weng, L.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016, 30, 2885–2898. [Google Scholar] [CrossRef]
- Naz, S.; Kolmert, J.; Yang, M.; Reinke, S.N.; Kamleh, M.A.; Snowden, S.; Heyder, T.; Levänen, B.; Erle, D.J.; Sköld, C.M.; et al. Metabolomics analysis identifies sex-associated metabotypes of oxidative stress and the autotaxin-lysoPA axis in COPD. Eur. Respir. J. 2017, 49, 1602322. [Google Scholar] [CrossRef]
- Li, Q.; Wong, W.R.; Chakrabarti, A.; Birnberg, A.; Yang, X.; Verschueren, E.; Neighbors, M.; Rosenberger, C.; Grimbaldeston, M.; Tew, G.W.; et al. Serum Lysophosphatidic Acid Measurement by Liquid Chromatography-Mass Spectrometry in COPD Patients. J. Am. Soc. Mass Spectrom. 2021, 32, 1987–1997. [Google Scholar] [CrossRef]
- Halper-Stromberg, E.; Gillenwater, L.; Cruickshank-Quinn, C.; O’Neal, W.K.; Reisdorph, N.; Petrache, I.; Zhuang, Y.; Labaki, W.W.; Curtis, J.L.; Wells, J.; et al. Bronchoalveolar Lavage Fluid from COPD Patients Reveals More Compounds Associated with Disease than Matched Plasma. Metabolites 2019, 9, 157. [Google Scholar] [CrossRef]
- Cruickshank-Quinn, C.I.; Jacobson, S.; Hughes, G.; Powell, R.L.; Petrache, I.; Kechris, K.; Bowler, R.; Reisdorph, N. Metabolomics and transcriptomics pathway approach reveals outcome-specific perturbations in COPD. Sci. Rep. 2018, 8, 17132. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, C.; Tian, S.; Wang, J.; Gai, X.; Zhou, Q.; Chen, Y.; Gao, X.; Sun, Y.; Liang, Y. Differences in the lipid metabolism profile and clinical characteristics between eosinophilic and non-eosinophilic acute exacerbation of chronic obstructive pulmonary disease. Front. Mol. Biosci. 2023, 10, 1204985. [Google Scholar] [CrossRef]
- Blom, M.; Tool, A.T.; Wever, P.C.; Wolbink, G.J.; Brouwer, M.C.; Calafat, J.; Egesten, A.; Knol, E.F.; Hack, C.E.; Roos, D.; et al. Human eosinophils express, relative to other circulating leukocytes, large amounts of secretory 14-kD phospholipase A2. Blood 1998, 91, 3037–3043. [Google Scholar]
- Nishiyama, O.; Kume, H.; Kondo, M.; Ito, Y.; Ito, M.; Yamaki, K. Role of lysophosphatidylcholine in eosinophil infiltration and resistance in airways. Clin. Exp. Pharmacol. Physiol. 2004, 31, 179–184. [Google Scholar] [CrossRef]
- Zhu, X.; Learoyd, J.; Butt, S.; Zhu, L.; Usatyuk, P.V.; Natarajan, V.; Munoz, N.M.; Leff, A.R. Regulation of eosinophil adhesion by lysophosphatidylcholine via a non-store-operated Ca2+ channel. Am. J. Respir. Cell Mol. Biol. 2007, 36, 585–593. [Google Scholar] [CrossRef]
- Gan, L.; Xue, J.X.; Li, X.; Liu, D.S.; Ge, Y.; Ni, P.Y.; Deng, L.; Lu, Y.; Jiang, W. Blockade of lysophosphatidic acid receptors LPAR1/3 ameliorates lung fibrosis induced by irradiation. Biochem. Biophys. Res. Commun. 2011, 409, 7–13. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Receptor | G-Protein Subunit | Downstream Signaling | Specific Biological Functions |
|---|---|---|---|
| LPAR1-6 (GPCR) | Gα12/13 | Rock, Rho/SRF | Cell motility, infiltration, cytoskeletal rearrangements |
| Gαq/11 | PLC, IP3 | Vasodilation, cell growth, immune responses | |
| Gαs | AC/cAMP | Production of cAMP | |
| Gαi/o | PLC, Ras/MAPK, PI3K/Rac, PI3K/Akt | Morphological alterations, cell migration, survival |
| Study | Sample Size | LPC Levels (COPD vs. Control) | LPA Levels (COPD vs. Control) | Correlations with Lung Function | Main Findings |
|---|---|---|---|---|---|
| Naz et al. (2017) [94] | Healthy smokers (n = 40) and COPD Patients (n = 38) | N/A | LPA (16:0) and LPA (18:2) were increased significantly (p < 0.05). | LPA (16:0) and LPA (18:2) were correlated with FEV1 in male COPD patients but not in females. | LPA (16:0) and LPA (18:2) were increased significantly in COPD patients and correlated with FEV1 in male COPD patients but not in females. |
| Li et al. (2021) [95] | Two cohorts of samples, a small cohort, healthy controls (n = 10) and COPD patients (n = 11). A large cohort, COPD patients (n = 268). | N/A | LPA(16:0), LPA(18:0), LPA(18:1), LPA(18:2) were increased significantly (p < 0.05). | No correlation with LPA and FEV1 or FEV1/FVC. | The correlation was not significant between LPA and FEV1, or LPA and postbronchodilator ratio FEV1/FVC in either female or male patients. |
| Li et al. (2021) [46] | COPD patients (n = 136) | N/A | N/A | N/A | Patients with low and intermediate levels of LPA (especially LPA (16:0) and LPA (20:4)) had a higher incidence of exacerbation than those with high LPA levels. |
| Cruickshank-Quinn et al. (2018) [97] | COPD patients (n = 149) | N/A | N/A | LPC (16:0) and LPC (18:1) were negatively correlated with the FEV1%pred and the ratio of FEV1/FVC | LPC (16:0) and LPC (18:1) were negatively correlated with the FEV1%pred and the ratio of FEV1/FVC |
| Eitan Halper-Stromberg et al. (2019) [96] | COPD Patients (n = 47) | N/A | N/A | No significant correlation between FEV1/FVC and LPC | There was no significant correlation between FEV1/FVC and LPC in BAL from patients with COPD. |
| Gai et al. (2022) [4] | AECOPD Patients (n = 58) | N/A | N/A | N/A | LPC (18:3) was significantly lower during acute exacerbations than that in recovery stage in COPD patients, especially in non-eosinophilic exacerbators. |
| Wang et al. (2023) [98] | AECOPD patients (n = 71) | N/A | N/A | N/A | LPC (16:0) and LPC (20:2) levels were increased in eosinophilic AECOPD and were associated with certain positive clinical outcomes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Chen, Y.; Liang, Y.; Sun, Y. The Role of Lysophospholipid Metabolites LPC and LPA in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Metabolites 2024, 14, 317. https://doi.org/10.3390/metabo14060317
Zhou Q, Chen Y, Liang Y, Sun Y. The Role of Lysophospholipid Metabolites LPC and LPA in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Metabolites. 2024; 14(6):317. https://doi.org/10.3390/metabo14060317
Chicago/Turabian StyleZhou, Qiqiang, Yahong Chen, Ying Liang, and Yongchang Sun. 2024. "The Role of Lysophospholipid Metabolites LPC and LPA in the Pathogenesis of Chronic Obstructive Pulmonary Disease" Metabolites 14, no. 6: 317. https://doi.org/10.3390/metabo14060317
APA StyleZhou, Q., Chen, Y., Liang, Y., & Sun, Y. (2024). The Role of Lysophospholipid Metabolites LPC and LPA in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Metabolites, 14(6), 317. https://doi.org/10.3390/metabo14060317