
Genetic Evidence for Causal Relationships between Plasma Eicosanoid Levels and Cardiovascular Disease

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Data Sources
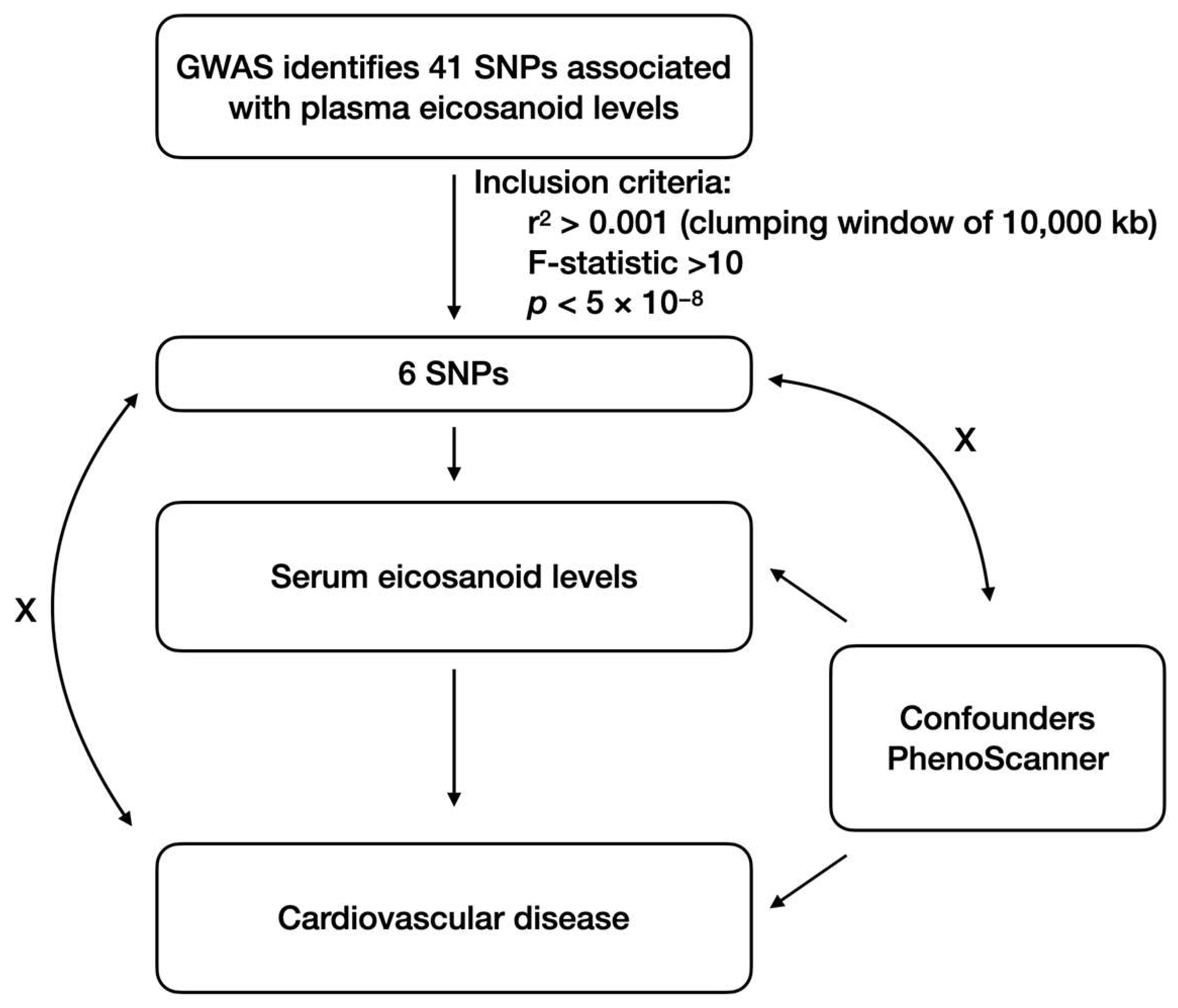
2.3. Selection of Genetic Instruments
2.4. MR Estimates
2.5. Sensitivity Analysis
3. Results
3.1. Characteristics of Selected Genetic Instruments
3.2. Causal Effects of Plasma Eicosanoid Levels on CVD
3.3. Sensitivity Analyses Did Not Display an Indication of Unknown Pleiotropy
4. Discussion
4.1. Eicosanoid and Ischemic CVD
4.2. Eicosanoid and HF
4.3. Eicosanoid and Pulmonary Embolism
4.4. Limitation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Writing Committee Members; Virani, S.S.; Newby, L.K.; Arnold, S.V.; Bittner, V.; Brewer, L.C.; Demeter, S.H.; Dixon, D.L.; Fearon, W.F.; Hess, B.; et al. 2023 Aha/Acc/Accp/Aspc/Nla/Pcna Guideline for the Management of Patients with Chronic Coronary Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2023, 82, 833–955. [Google Scholar]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary Pufas. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bi, X. Integrated Control of Fatty Acid Metabolism in Heart Failure. Metabolites 2023, 13, 615. [Google Scholar] [CrossRef]
- Conway, M.C.; McSorley, E.M.; Mulhern, M.S.; Strain, J.J.; van Wijngaarden, E.; Yeates, A.J. Influence of Fatty Acid Desaturase (Fads) Genotype on Maternal and Child Polyunsaturated Fatty Acids (Pufa) Status and Child Health Outcomes: A Systematic Review. Nutr. Rev. 2020, 78, 627–646. [Google Scholar] [CrossRef]
- Brocker, C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Evolutionary Divergence and Functions of the Human Acyl-Coa Thioesterase Gene (ACOT) Family. Hum. Genom. 2010, 4, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-Coa Metabolism and Partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. Eicosanoids and Cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Soodi, D.; VanWormer, J.J.; Rezkalla, S.H. Aspirin in Primary Prevention of Cardiovascular Events. Clin. Med. Res. 2020, 18, 89–94. [Google Scholar] [CrossRef]
- Antman, E.M.; DeMets, D.; Loscalzo, J. Cyclooxygenase Inhibition and Cardiovascular Risk. Circulation 2005, 112, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Lüscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Wang, D.W. The Roles of Eicosanoids in Myocardial Diseases. Adv. Pharmacol. 2023, 97, 167–200. [Google Scholar] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-Inflammatory Properties of Cytochrome P450 Epoxygenase-Derived Eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.B. New Role for Epoxyeicosatrienoic Acids as Anti-Inflammatory Mediators. Trends Pharmacol. Sci. 2000, 21, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.B.; Gebremedhin, D.; Pratt, P.F.; Harder, D.R. Identification of Epoxyeicosatrienoic Acids as Endothelium-Derived Hyperpolarizing Factors. Circ. Res. 1996, 78, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Holla, V.R.; Adas, F.; Imig, J.D.; Zhao, X.; Price, E., Jr.; Olsen, N.; Kovacs, W.J.; Magnuson, M.A.; Keeney, D.S.; Breyer, M.D.; et al. Alterations in the Regulation of Androgen-Sensitive Cyp 4a Monooxygenases Cause Hypertension. Proc. Natl. Acad. Sci. USA 2001, 98, 5211–5216. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Holla, V.R.; Wei, Y.; Wang, W.H.; Gatica, A.; Wei, S.; Mei, S.; Miller, C.M.; Cha, D.R.; Price, E.; et al. Salt-Sensitive Hypertension Is Associated with Dysfunctional Cyp4a10 Gene and Kidney Epithelial Sodium Channel. J. Clin. Investig. 2006, 116, 1696–1702. [Google Scholar] [CrossRef]
- Imig, J.D. Orally Active Epoxyeicosatrienoic Acid Analogs in Hypertension and Renal Injury. Adv. Pharmacol. 2022, 94, 27–55. [Google Scholar] [PubMed]
- Rhee, E.P.; Surapaneni, A.L.; Schlosser, P.; Alotaibi, M.; Yang, Y.-N.; Coresh, J.; Jain, M.; Cheng, S.; Yu, B.; Grams, M.E. A Genome-Wide Association Study Identifies 41 Loci Associated with Eicosanoid Levels. Commun. Biol. 2023, 6, 792. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A Cross-Population Atlas of Genetic Associations for 220 Human Phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. Finngen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Hartiala, J.A.; Han, Y.; Jia, Q.; Hilser, J.R.; Huang, P.; Gukasyan, J.; Schwartzman, W.S.; Cai, Z.; Biswas, S.; Tregouet, D.-A.; et al. Genome-Wide Analysis Identifies Novel Susceptibility Loci for Myocardial Infarction. Eur. Heart J. 2021, 42, 919–933. [Google Scholar] [CrossRef]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.K.; Van Der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry Genome-Wide Association Study of 520,000 Subjects Identifies 32 Loci Associated with Stroke and Stroke Subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef]
- Shah, S.; Henry, A.; Roselli, C.; Lin, H.; Sveinbjornsson, G.; Fatemifar, G.; Hedman, A.K.; Wilk, J.B.; Morley, M.P.; Chaffin, M.D.; et al. Genome-Wide Association and Mendelian Randomisation Analysis Provide Insights into the Pathogenesis of Heart Failure. Nat. Commun. 2020, 11, 163. [Google Scholar] [CrossRef]
- Fadista, J.; Manning, A.K.; Florez, J.C.; Groop, L. The (in)Famous Gwas P-Value Threshold Revisited and Updated for Low-Frequency Variants. Eur. J. Hum. Genet. 2016, 24, 1202–1205. [Google Scholar] [CrossRef]
- Palmer, T.M.; Lawlor, D.A.; Harbord, R.M.; Sheehan, N.A.; Tobias, J.H.; Timpson, N.J.; Smith, G.D.; Sterne, J.A. Using Multiple Genetic Variants as Instrumental Variables for Modifiable Risk Factors. Stat. Methods Med. Res. 2011, 21, 223–242. [Google Scholar] [CrossRef]
- Burgess, S.; Scott, R.A.; Timpson, N.J.; Smith, G.D.; Thompson, S.G.; EPIC-InterAct Consortium. Using Published Data in Mendelian Randomization: A Blueprint for Efficient Identification of Causal Risk Factors. Eur. J. Epidemiol. 2015, 30, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, F.P.; Smith, G.D.; Bowden, J. Robust Inference in Summary Data Mendelian Randomization Via the Zero Modal Pleiotropy Assumption. Leuk. Res. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.C.; Timpson, N.; Smith, G.D. Mendelian Randomization: Using Genes as Instruments for Making Causal Inferences in Epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-J.; Tan, J.-S.; Gao, X.-J.; Yang, J.-G.; Yang, Y.-J. Effect of Cheese Intake on Cardiovascular Diseases and Cardiovascular Biomarkers. Nutrients 2022, 14, 2936. [Google Scholar] [CrossRef] [PubMed]
- Wootton, R.E.; Lawn, R.B.; Millard, L.A.C.; Davies, N.M.; Taylor, A.E.; Munafo, M.R.; Timpson, N.J.; Davis, O.S.P.; Smith, G.D.; Haworth, C.M.A. Evaluation of the Causal Effects between Subjective Wellbeing and Cardiometabolic Health: Mendelian Randomisation Study. BMJ 2018, 362, k3788. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Bowden, J.; Fall, T.; Ingelsson, E.; Thompson, S.G. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology 2017, 28, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Staley, J.R.; Blackshaw, J.; Kamat, M.A.; Ellis, S.; Surendran, P.; Sun, B.B.; Paul, D.S.; Freitag, D.; Burgess, S.; Danesh, J.; et al. Phenoscanner: A Database of Human Genotype-Phenotype Associations. Bioinformatics 2016, 32, 3207–3209. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cheng, J.; Wang, Y. Genetic Support of a Causal Relationship between Iron Status and Atrial Fibrillation: A Mendelian Randomization Study. Genes Nutr. 2022, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dai, Z.; Guo, R.; Wang, X.; Gong, W.; Duan, J.; He, Z.; Ding, R.; Zhang, X.; Nie, S.; et al. Metabolomics Reveal Dynamic Changes in Eicosanoid Profile in Patients with St-Elevation Myocardial Infarction after Percutaneous Coronary Intervention. Clin. Exp. Pharmacol. Physiol. 2021, 48, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Jugdutt, B.I. Prostaglandins in Myocardial: With Emphasis on Myocardial Preservation. Prostaglandins Med. 1981, 7, 109–123. [Google Scholar] [CrossRef]
- Schrör, K.; Thiemermann, C. Treatment of Acute Myocardial Ischaemia with a Selective Antagonist of Thromboxane Receptors (Bm 13.177). Br. J. Pharmacol. 1986, 87, 631–637. [Google Scholar] [CrossRef]
- Johnson, G., 3rd; Furlan, L.E.; Aoki, N.; Lefer, A.M. Endothelium and Myocardial Protecting Actions of Taprostene, a Stable Prostacyclin Analogue, after Acute Myocardial Ischemia and Reperfusion in Cats. Circ. Res. 1990, 66, 1362–1370. [Google Scholar] [CrossRef]
- Yokomizo, T.; Nakamura, M.; Shimizu, T. Leukotriene Receptors as Potential Therapeutic Targets. J. Clin. Investig. 2018, 128, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Hansson, G.K. Leukotriene Receptors in Atherosclerosis. Ann. Med. 2006, 38, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Spanbroek, R.; Gräbner, R.; Lötzer, K.; Hildner, M.; Urbach, A.; Rühling, K.; Moos, M.P.W.; Kaiser, B.; Cohnert, T.U.; Wahlers, T.; et al. Expanding Expression of the 5-Lipoxygenase Pathway within the Arterial Wall during Human Atherogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.; Hermansson, A.; Hlawaty, H.; Letourneur, D.; Yan, Z.Q.; Bäck, M. The Leukotriene B4 Receptor (Blt) Antagonist Biil284 Decreases Atherosclerosis in Apoe−/− Mice. Prostaglandins Other Lipid Mediat. 2015, 121 Pt A, 105–109. [Google Scholar] [CrossRef]
- de Hoog, V.C.; Bovens, S.M.; de Jager, S.C.; van Middelaar, B.J.; van Duijvenvoorde, A.; Doevendans, P.A.; Pasterkamp, G.; de Kleijn, D.P.; Timmers, L. Blt1 Antagonist Lsn2792613 Reduces Infarct Size in a Mouse Model of Myocardial Ischaemia-Reperfusion Injury. Cardiovasc. Res. 2015, 108, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, T.; He, X.; Liu, X.; Wang, B.; Liu, Y.; Li, Z.; Tan, R.; Ding, C.; Wang, H.; et al. Cyp2j2 Overexpression Increases Eets and Protects against Hfd-Induced Atherosclerosis in Apoe−/− Mice. J. Cardiovasc. Pharmacol. 2016, 67, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Seubert, J.; Yang, B.; Bradbury, J.A.; Graves, J.; Degraff, L.M.; Gabel, S.; Gooch, R.; Foley, J.; Newman, J.; Mao, L.; et al. Enhanced Postischemic Functional Recovery in Cyp2j2 Transgenic Hearts Involves Mitochondrial Atp-Sensitive K+ Channels and P42/P44 Mapk Pathway. Circ. Res. 2004, 95, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Huang, J.; Wang, D.; Chen, L.; Sun, D.; Zhao, C. Endothelium-Specific Cyp2j2 Overexpression Improves Cardiac Dysfunction by Promoting Angiogenesis Via Jagged1/Notch1 Signaling. J. Mol. Cell. Cardiol. 2018, 123, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.J.; Hsu, A.; Falck, J.R.; Nithipatikom, K. Mechanisms by Which Epoxyeicosatrienoic Acids (Eets) Elicit Cardioprotection in Rat Hearts. J. Mol. Cell. Cardiol. 2007, 42, 687–691. [Google Scholar] [CrossRef]
- Wu, C.-C.; Mei, S.; Cheng, J.; Ding, Y.; Weidenhammer, A.; Garcia, V.; Zhang, F.; Gotlinger, K.; Manthati, V.L.; Falck, J.R.; et al. Androgen-Sensitive Hypertension Associates with Upregulated Vascular CYP4A12–20-HETE Synthase. J. Am. Soc. Nephrol. 2013, 24, 1288–1296. [Google Scholar] [CrossRef]
- Shekhar, S.; Varghese, K.; Li, M.; Fan, L.; Booz, G.W.; Roman, R.J.; Fan, F. Conflicting Roles of 20-Hete in Hypertension and Stroke. Int. J. Mol. Sci. 2019, 20, 4500. [Google Scholar] [CrossRef]
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar]
- Lau, E.S.; Roshandelpoor, A.; Zarbafian, S.; Wang, D.; Guseh, J.S.; Allen, N.; Varadarajan, V.; Nayor, M.; Shah, R.V.; Lima, J.A.; et al. Eicosanoid and Eicosanoid-Related Inflammatory Mediators and Exercise Intolerance in Heart Failure with Preserved Ejection Fraction. Nat. Commun. 2023, 14, 7557. [Google Scholar] [CrossRef] [PubMed]
- Kayama, Y.; Minamino, T.; Toko, H.; Sakamoto, M.; Shimizu, I.; Takahashi, H.; Okada, S.; Tateno, K.; Moriya, J.; Yokoyama, M. Cardiac 12/15 Lipoxygenase-Induced Inflammation Is Involved in Heart Failure. J. Exp. Med. 2009, 206, 1565–1574. [Google Scholar] [CrossRef]
- Kain, V.; Ingle, K.A.; Kabarowski, J.; Barnes, S.; Limdi, N.A.; Prabhu, S.D.; Halade, G.V. Genetic Deletion of 12/15 Lipoxygenase Promotes Effective Resolution of Inflammation Following Myocardial Infarction. J. Mol. Cell. Cardiol. 2018, 118, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Kala, P.; Hnat, T.; Padrova, K.; Kotaška, K.; Veselka, J. Eicosanoids in Human Heart Failure: Pilot Study of Plasma Epoxyeicosatrienoic and Dihydroxyeicosatrienoic Acid Levels. Arch. Med. Sci. 2023, 19, 513–517. [Google Scholar] [CrossRef]
- He, Z.; Zhang, X.; Chen, C.; Wen, Z.; Hoopes, S.L.; Zeldin, D.C.; Wang, D.W. Cardiomyocyte-Specific Expression of Cyp2j2 Prevents Development of Cardiac Remodelling Induced by Angiotensin Ii. Cardiovasc. Res. 2015, 105, 304–317. [Google Scholar] [CrossRef]
- He, Z.; Yang, Y.; Wen, Z.; Chen, C.; Xu, X.; Zhu, Y.; Wang, Y.; Wang, D.W. Cyp2j2 Metabolites, Epoxyeicosatrienoic Acids, Attenuate Ang Ii-Induced Cardiac Fibrotic Response by Targeting Galpha(12/13). J. Lipid Res. 2017, 58, 1338–1353. [Google Scholar] [CrossRef]
- Freund, Y.; Cohen-Aubart, F.; Bloom, B. Acute Pulmonary Embolism: A Review. JAMA 2022, 328, 1336–1345. [Google Scholar] [CrossRef]
- Ikdahl, E.; Rollefstad, S.; Kazemi, A.; Provan, S.A.; Larsen, T.L.; Semb, A.G. Non-Steroidal Anti-Inflammatory Drugs and Risk of Pulmonary Embolism in Patients with Inflammatory Joint Disease-Results from the Nationwide Norwegian Cardio-Rheuma Registry. Eur. Heart J. Cardiovasc. Pharmacother. 2024, 10, 27–34. [Google Scholar] [CrossRef]
- Utsunomiya, T.; Krausz, M.M.; Levine, L.; Shepro, D.; Hechtman, H.B. Thromboxane Mediation of Cardiopulmonary Effects of Embolism. J. Clin. Investig. 1982, 70, 361–368. [Google Scholar] [CrossRef]
- Perlman, M.B.; Johnson, A.; Jubiz, W.; Malik, A.B. Lipoxygenase Products Induce Neutrophil Activation and Increase Endothelial Permeability after Thrombin-Induced Pulmonary Microembolism. Circ. Res. 1989, 64, 62–73. [Google Scholar] [CrossRef]
- Shen, T.; Shi, J.; Wang, N.; Yu, X.; Zhang, C.; Li, J.; Wei, L.; Ma, C.; Zhao, X.; Lian, M.; et al. 15-Lipoxygenase and 15-Hydroxyeicosatetraenoic Acid Regulate Intravascular Thrombosis in Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L449–L462. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Locus | SNP | Chromosome | Position | Effect_Allele | Other_ Allele | Beta | Se | p-Value | Eaf | F |
|---|---|---|---|---|---|---|---|---|---|---|
| FADS1-3 | rs174544 | 11 | 61800281 | C | A | 0.54 | 0.018 | 2.76 × 10−196 | 0.29 | 65 |
| SLCO1B1 | rs4149056 | 12 | 21178615 | T | C | 0.54 | 0.023 | 5.7 × 10−120 | 0.16 | 25 |
| PKD2L1 | rs603424 | 10 | 100315722 | G | A | −0.41 | 0.022 | 6.12 × 10−74 | 0.18 | 17 |
| ACOT4/ACOT6 | rs111511359 | 14 | 73610482 | G | T | −0.34 | 0.021 | 7.68 × 10−59 | 0.21 | 15 |
| ACSM6 | rs612490 | 10 | 95215869 | A | G | −0.23 | 0.018 | 1.92 × 10−38 | 0.57 | 14 |
| CYP11B1/CYP11B2 | rs4736317 | 8 | 142901337 | A | G | −0.20 | 0.018 | 6.37 × 10−30 | 0.56 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, X.; Wang, Y.; Lin, Y.; Wang, M.; Li, X. Genetic Evidence for Causal Relationships between Plasma Eicosanoid Levels and Cardiovascular Disease. Metabolites 2024, 14, 294. https://doi.org/10.3390/metabo14060294
Bi X, Wang Y, Lin Y, Wang M, Li X. Genetic Evidence for Causal Relationships between Plasma Eicosanoid Levels and Cardiovascular Disease. Metabolites. 2024; 14(6):294. https://doi.org/10.3390/metabo14060294
Chicago/Turabian StyleBi, Xukun, Yiran Wang, Yangjun Lin, Meihui Wang, and Xiaoting Li. 2024. "Genetic Evidence for Causal Relationships between Plasma Eicosanoid Levels and Cardiovascular Disease" Metabolites 14, no. 6: 294. https://doi.org/10.3390/metabo14060294
APA StyleBi, X., Wang, Y., Lin, Y., Wang, M., & Li, X. (2024). Genetic Evidence for Causal Relationships between Plasma Eicosanoid Levels and Cardiovascular Disease. Metabolites, 14(6), 294. https://doi.org/10.3390/metabo14060294