
Metabolomics Profiling of Stages of Coronary Artery Disease Progression

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. The Study Population
2.2. Sample Collection and Processing
2.3. Biochemical and Biometric Analysis
2.4. Metabolomics Profiling
2.5. Statistical Analysis
3. Results
3.1. Characteristics of Participants
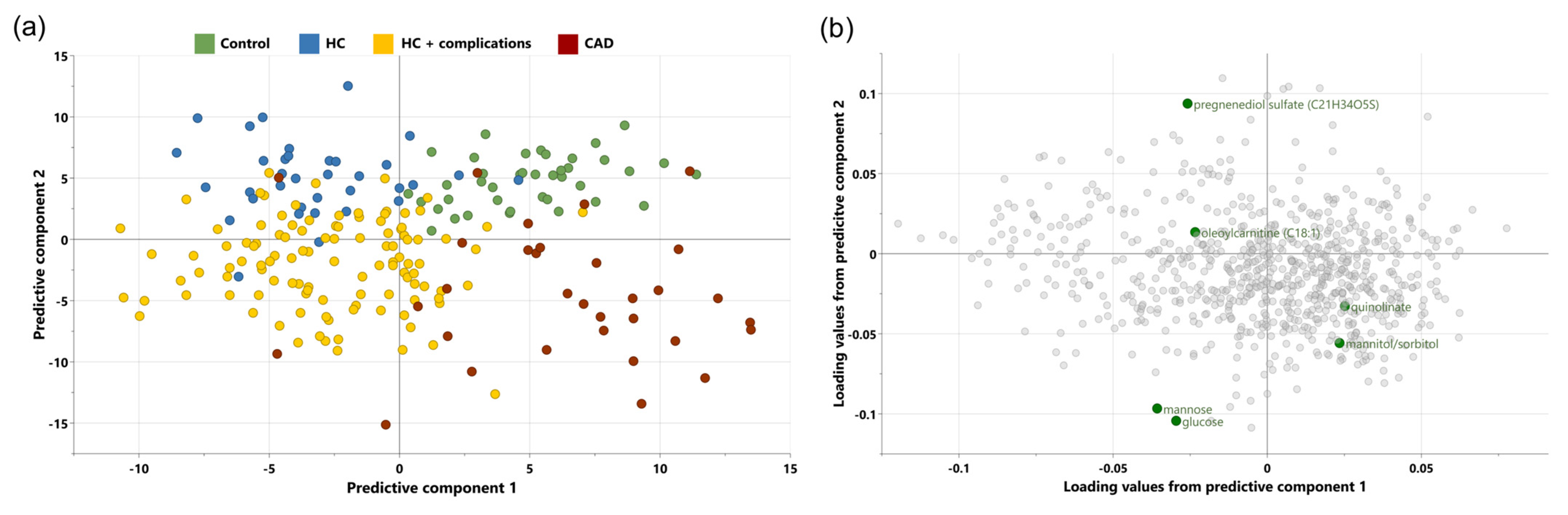
3.2. Multivariate Analysis
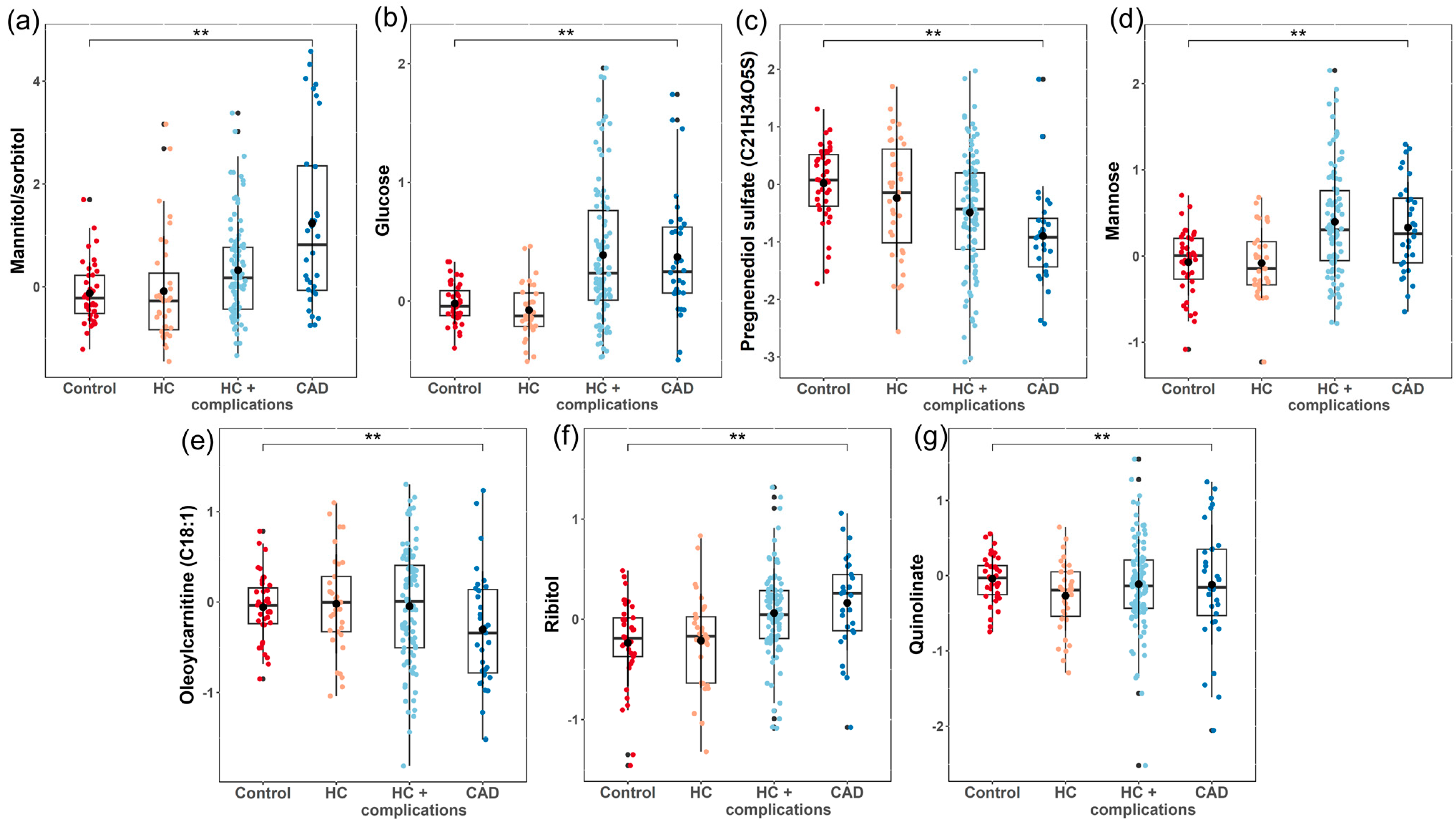
3.3. Univariate Analysis
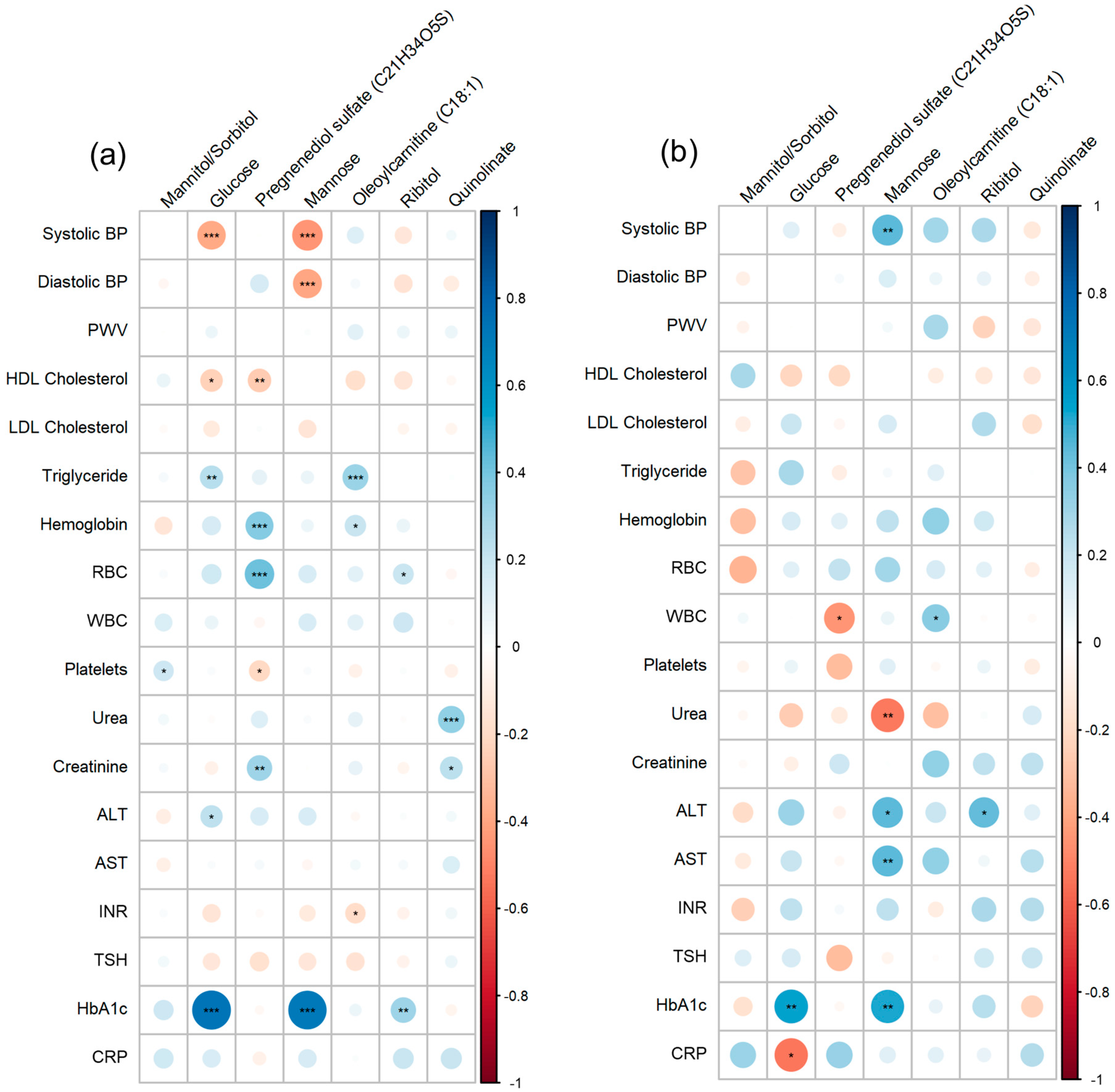
3.4. Correlation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Mechanisms of Acute Coronary Syndromes and Their Implications for Therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement fromthe European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Robinson, J.G.; Brook, R.D.; Catapano, A.L.; Chapman, M.J.; Neff, D.R.; Voros, S.; Giugliano, R.P.; Smith, G.D.; Fazio, S.; et al. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N. Engl. J. Med. 2016, 375, 2144–2153. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Juutilainen, A.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Type 2 diabetes as a “coronary heart disease equivalent”: An 18-year prospective population-based study in Finnish subjects. Diabetes Care 2005, 28, 2901–2907. Available online: https://pubmed.ncbi.nlm.nih.gov/16306552/ (accessed on 28 April 2023). [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011, 14, 575–585. [Google Scholar] [CrossRef]
- Brownlee, M. The Pathobiology of Diabetic ComplicationsA Unifying Mechanism. Diabetes 2005, 54, 1615–1625. Available online: https://diabetesjournals.org/diabetes/article/54/6/1615/14015/The-Pathobiology-of-Diabetic-ComplicationsA (accessed on 28 April 2023). [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Coca, A.; Dominiczak, A.; Kahan, T.; Mahfoud, F.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Kardiol. Pol. 2019, 77, 71–159. [Google Scholar] [CrossRef]
- Vasan, R.S.; Beiser, A.; Seshadri, S.; Larson, M.G.; Kannel, W.B.; D’Agostino, R.B.; Levy, D. Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham Heart Study. JAMA 2002, 287, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, N.; Gao, P.; Kondapally Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [PubMed]
- Zhu, X.; Yu, L.; Zhou, H.; Ma, Q.; Zhou, X.; Lei, T.; Hu, J.; Xu, W.; Yi, N.; Lei, S. Atherogenic index of plasma is a novel and better biomarker associated with obesity: A population-based cross-sectional study in China. Lipids Health Dis. 2018, 17, 37. Available online: https://lipidworld.biomedcentral.com/articles/10.1186/s12944-018-0686-8 (accessed on 9 May 2023). [CrossRef]
- Al Shawaf, E.; Al-Ozairi, E.; Al-Asfar, F.; Mohammad, A.; Al-Beloushi, S.; Devarajan, S.; Al-Mulla, F.; Abubaker, J.; Arefanian, H. Atherogenic Index of Plasma (AIP) a Tool to Assess Changes in Cardiovascular Disease Risk Post Laparoscopic Sleeve Gastrectomy. J. Diabetes Res. 2020, 2020, 2091341. [Google Scholar] [CrossRef]
- Li, Y.W.; Kao, T.W.; Chang, P.K.; Chen, W.L.; Wu, L.W. Atherogenic index of plasma as predictors for metabolic syndrome, hypertension and diabetes mellitus in Taiwan citizens: A 9-year longitudinal study. Sci. Rep. 2021, 11, 9900. Available online: https://www.nature.com/articles/s41598-021-89307-z (accessed on 9 May 2023). [CrossRef]
- Sajdeya, O.; Beran, A.; Mhanna, M.; Alharbi, A.; Burmeister, C.; Abuhelwa, Z.; Malhas, S.-E.; Khader, Y.; Sayeh, W.; Assaly, R.; et al. Triglyceride Glucose Index for the Prediction of Subclinical Atherosclerosis and Arterial Stiffness: A Meta-analysis of 37,780 Individuals. Curr. Probl. Cardiol. 2022, 47, 101390. [Google Scholar] [CrossRef] [PubMed]
- Mangalesh, S.; Yadav, P.; Dudani, S.; Mahesh, N.K. Atherogenic index of plasma predicts coronary artery disease severity and major adverse cardiac events in absence of conventional risk factors. Coron. Artery Dis. 2022, 33, 523–530. Available online: https://journals.lww.com/coronary-artery/Fulltext/2022/11000/Atherogenic_index_of_plasma_predicts_coronary.2.aspx (accessed on 9 May 2023). [CrossRef]
- Cheng, S.; Shah, S.H.; Corwin, E.J.; Fiehn, O.; Fitzgerald, R.L.; Gerszten, R.E.; Illig, T.; Rhee, E.P.; Srinivas, P.R.; Wang, T.J.; et al. Potential Impact and Study Considerations of Metabolomics in Cardiovascular Health and Disease: A Scientific Statement from the American Heart Association. Circ. Cardiovasc. Genet. 2017, 10, e000032. [Google Scholar] [CrossRef]
- Al Kuwari, H.; Al Thani, A.; Al Marri, A.; Al Kaabi, A.; Abderrahim, H.; Afifi, N.; Qafoud, F.; Chan, Q.; Tzoulaki, I.; Downey, P.; et al. The Qatar Biobank: Background and methods. BMC Public Health 2015, 15, 1208. [Google Scholar] [CrossRef]
- Al-Khelaifi, F.; Diboun, I.; Donati, F.; Botrè, F.; Alsayrafi, M.; Georgakopoulos, C.; Suhre, K.; Yousri, N.A.; Elrayess, M.A. A pilot study comparing the metabolic profiles of elite-level athletes from different sporting disciplines. Sports Med. Open 2018, 4, 2. [Google Scholar] [CrossRef]
- D’Agostino Sr, R.B.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham heart study. Circulation 2008, 117, 743–753. Available online: http://circ.ahajournals.org (accessed on 28 December 2023). [CrossRef] [PubMed]
- Kattoor, A.J.; Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. Available online: https://pubmed.ncbi.nlm.nih.gov/29737246/ (accessed on 16 April 2023). [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K.L.; Söderberg-Nauclér, C. Inflammation and Atherosclerosis. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 297–329. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclenrosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Rudijanto, A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med. Indones 2007, 39, 86–93. Available online: https://pubmed.ncbi.nlm.nih.gov/17933075/ (accessed on 14 April 2023). [PubMed]
- Qi, X.; Tester, R.F. Fructose, galactose and glucose—In health and disease. Clin. Nutr. Espen. 2019, 33, 18–28. Available online: https://pubmed.ncbi.nlm.nih.gov/31451258/ (accessed on 1 May 2023). [CrossRef]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Mirtschink, P.; Jang, C.; Arany, Z.; Krek, W. Fructose metabolism, cardiometabolic risk, and the epidemic of coronary artery disease. Eur. Heart J. 2017, 39, 2497–2505. [Google Scholar] [CrossRef]
- Kolderup, A.; Svihus, B. Fructose Metabolism and Relation to Atherosclerosis, Type 2 Diabetes, and Obesity. J. Nutr. Metab. 2015, 2015, 823081. [Google Scholar] [CrossRef]
- Frayn, K.N.; Kingman, S.M. Dietary sugars and lipid metabolism in humans. Am. J. Clin. Nutr. 1995, 62, 250S–263S. Available online: https://academic.oup.com/ajcn/article/62/1/250S/4651719 (accessed on 3 May 2023). [CrossRef] [PubMed]
- Alam, Y.H.; Kim, R.; Jang, C. Metabolism and Health Impacts of Dietary Sugars. J. Lipid Atheroscler. 2022, 11, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Fortin, E.; Ferrannini, G.; Campi, B.; Mellbin, L.; Norhammar, A.; Näsman, P.; Saba, A.; Ferrannini, E.; Rydén, L. Plasma mannose as a novel marker of myocardial infarction across different glycaemic states: A case control study. Cardiovasc. Diabetol. 2022, 21, 195. [Google Scholar] [CrossRef] [PubMed]
- Howard, B.V.; Wylie-Rosett, J. Sugar and Cardiovascular Disease. Circulation 2002, 106, 523–527. [Google Scholar] [CrossRef]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The diabetes mellitus–atherosclerosis connection: The role of lipid and glucose metabolism and chronic inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: From pharmacology to pre-clinical and clinical therapeutics. Theranostics 2021, 11, 4502–4515. [Google Scholar] [CrossRef]
- Yamagishi, S.I.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 663–671. [Google Scholar] [CrossRef]
- Yamagishi, S.I.; Nakamura, N.; Suematsu, M.; Kaseda, K.; Matsui, T. Advanced glycation end products: A molecular target for vascular complications in diabetes. Mol. Med. 2015, 21, S32–S40. [Google Scholar] [CrossRef]
- Cagnina, A.; Chabot, O.; Davin, L.; Lempereur, M.; Maréchal, P.; Oury, C.; Lancellotti, P. Atherosclerosis, an inflammatory disease. Rev. Medicale Liege 2022, 77, 302–309. [Google Scholar]
- Nakagami, H.; Kaneda, Y.; Ogihara, T.; Morishita, R. Endothelial Dysfunction in Hyperglycemia as a Trigger of Atherosclerosis. Curr. Diabetes Rev. 2005, 1, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Silambarasan, M.; Tan, J.R.; Karolina, D.S.; Armugam, A.; Kaur, C.; Jeyaseelan, K. MicroRNAs in hyperglycemia induced endothelial cell dysfunction. Int. J. Mol. Sci. 2016, 17, 518. [Google Scholar] [CrossRef]
- Ferroni, P.; Basili, S.; Falco, A.; Davì, G. Platelet activation in type 2 diabetes mellitus. J. Thromb. Haemost. 2004, 2, 1282–1291. [Google Scholar] [CrossRef]
- Wang, L.; Yan, N.; Zhang, M.; Pan, R.; Dang, Y.; Niu, Y. The association between blood glucose levels and lipids or lipid ratios in type 2 diabetes patients: A cross-sectional study. Front. Endocrinol. 2022, 13, 969080. [Google Scholar] [CrossRef] [PubMed]
- Terashita, D.; Sawada, T.; Shiotani, H.; Nagasawa, Y.; Mizuguchi, T.; Okamoto, H.; Kim, S.; Koide, M.; Takarada, A. Ameliorating effects of miglitol on postprandial hyperglycemia and triglyceride/HDL ratio are associated with beneficial impact on atherosclerosis in diabetic patients with coronary artery disease. Eur. Heart J. 2013, 34 (Suppl. S1), P2271. [Google Scholar] [CrossRef]
- Pierce, S.R.; Germann, A.L.; Steinbach, J.H.; Akk, G. The Sulfated Steroids Pregnenolone Sulfate and Dehydroepiandrosterone Sulfate Inhibit the α 1 β 3 γ 2L GABAA Receptor by Stabilizing a Novel Nonconducting State. Mol. Pharmacol. 2022, 101, 68–77. Available online: https://pubmed.ncbi.nlm.nih.gov/34853153/ (accessed on 3 May 2023). [CrossRef]
- Moffett, S.X.; Klein, E.A.; Brannigan, G.; Martin, J.V. L-3,3′,5-triiodothyronine and pregnenolone sulfate inhibit Torpedo nicotinic acetylcholine receptors. PLoS ONE 2019, 14, e0223272. [Google Scholar] [CrossRef] [PubMed]
- Krausova, B.H.; Kysilov, B.; Cerny, J.; Vyklicky, V.; Smejkalova, T.; Ladislav, M.; Balik, A.; Korinek, M.; Chodounska, H.; Kudova, E.; et al. Site of Action of Brain Neurosteroid Pregnenolone Sulfate at the N-Methyl-D-Aspartate Receptor. J. Neurosci. 2020, 40, 5922–5936. Available online: https://pubmed.ncbi.nlm.nih.gov/32611707/ (accessed on 3 May 2023). [CrossRef]
- Rege, J.; Nanba, A.T.; Auchus, R.J.; Ren, J.; Peng, H.-M.; Rainey, W.E.; Turcu, A.F. Adrenocorticotropin Acutely Regulates Pregnenolone Sulfate Production by the Human Adrenal In Vivo and In Vitro. Clin. Endocrinol. Metab. 2017, 103, 320–327. Available online: https://pubmed.ncbi.nlm.nih.gov/29126147/ (accessed on 3 May 2023). [CrossRef]
- Ieko, T.; Sasaki, H.; Maeda, N.; Fujiki, J.; Iwano, H.; Yokota, H. Analysis of Corticosterone and Testosterone Synthesis in Rat Salivary Gland Homogenates. Front. Endocrinol. 2019, 10, 479. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.M.; Collins, P. Role of Testosterone in the Treatment of Cardiovascular Disease. Eur. Cardiol. Rev. 2017, 12, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Goodale, T.; Sadhu, A.; Petak, S.; Robbins, R. Testosterone and the Heart. Methodist Debakey Cardiovasc. J. 2017, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Erdoğan, A.; Çakmak, E.Ö.; Güler, A.; Kılıçgedik, A.; Kırma, C. The Relationship Between Levels of Sex Steroids and Coronary Collateral Circulation in Men Patients with Coronary Artery Disease. Koşuyolu Heart J. 2022, 25, 33–39. [Google Scholar] [CrossRef]
- Joury, A.; Alshehri, M.; Li, L.Z.; Rezan, T. Androgenic steroids dysregulation and the risk of coronary artery disease. Expert Rev. Cardiovasc. Ther. 2022, 20, 343–349. Available online: https://pubmed.ncbi.nlm.nih.gov/35583488/ (accessed on 3 May 2023). [CrossRef] [PubMed]
- Yeh, J.J.; Lin, C.L.; Hsu, N.H.; Kao, C.H. Effects of statins and steroids on coronary artery disease and stroke in patients with interstitial lung disease and pulmonary fibrosis: A general population study. PLoS ONE 2021, 16, e0259153. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.; Fall, T.; Ärnlöv, J.; Elmståhl, S.; Sundström, J. Large-Scale Metabolomics and the Incidence of Cardiovascular Disease. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2023, 12, 26885. [Google Scholar] [CrossRef] [PubMed]
- Oleoylcarnitine|C25H47NO4—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Oleoylcarnitine (accessed on 9 May 2023).
- Tonazzi, A.; Giangregorio, N.; Console, L.; Palmieri, F.; Indiveri, C. The Mitochondrial Carnitine Acyl-carnitine Carrier (SLC25A20): Molecular Mechanisms of Transport, Role in Redox Sensing and Interaction with Drugs. Biomolecules 2021, 11, 521. [Google Scholar] [CrossRef]
- Dambrova, M.; Makrecka-Kuka, M.; Kuka, J.; Vilskersts, R.; Nordberg, D.; Attwood, M.M.; Smesny, S.; Zheng, J.; Wishart, D.S.; Liepinsh, E.; et al. Acylcarnitines: Nomenclature, Biomarkers, Therapeutic Potential, Drug Targets, and Clinical Trials. Pharmacol. Rev. 2022, 74, 506–551. Available online: https://pubmed.ncbi.nlm.nih.gov/35710135/ (accessed on 9 May 2023). [CrossRef]
- Deda, O.; Panteris, E.; Meikopoulos, T.; Begou, O.; Mouskeftara, T.; Karagiannidis, E.; Papazoglou, A.S.; Sianos, G.; Theodoridis, G.; Gika, H. Correlation of Serum Acylcarnitines with Clinical Presentation and Severity of Coronary Artery Disease. Biomolecules 2022, 12, 354. [Google Scholar] [CrossRef]
- Strand, E.; Pedersen, E.R.; Svingen, G.F.T.; Olsen, T.; Bjørndal, B.; Karlsson, T.; Dierkes, J.; Njølstad, P.R.; Mellgren, G.; Tell, G.S.; et al. Serum Acylcarnitines and Risk of Cardiovascular Death and Acute Myocardial Infarction in Patients with Stable Angina Pectoris. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2017, 6, e003620. [Google Scholar] [CrossRef]
- Zhao, S.; Feng, X.-F.; Huang, T.; Luo, H.-H.; Chen, J.-X.; Zeng, J.; Gu, M.; Li, J.; Sun, X.-Y.; Sun, D.; et al. The Association Between Acylcarnitine Metabolites and Cardiovascular Disease in Chinese Patients With Type 2 Diabetes Mellitus. Front. Endocrinol. 2020, 11, 212. [Google Scholar] [CrossRef]
- Carluccio, M.A.; Massaro, M.; Bonfrate, C.; Siculella, L.; Maffia, M.; Nicolardi, G.; Distante, A.; Storelli, C.; De Caterina, R. Oleic acid inhibits endothelial activation: A direct vascular antiatherogenic mechanism of a nutritional component in the Mediterranean diet. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 220–228. [Google Scholar] [CrossRef]
- Massaro, M.; Carluccio, M.A.; De Caterina, R. Direct vascular antiatherogenic effects of oleic acid: A clue to the cardioprotective effects of the Mediterranean diet. Cardiologia 1999, 44, 507–513. [Google Scholar] [PubMed]
- Yang, Z.-H.; Nill, K.; Takechi-Haraya, Y.; Playford, M.P.; Nguyen, D.; Yu, Z.-X.; Pryor, M.; Tang, J.; Rojulpote, K.V.; Mehta, N.N.; et al. Differential Effect of Dietary Supplementation with a Soybean Oil Enriched in Oleic Acid versus Linoleic Acid on Plasma Lipids and Atherosclerosis in LDLR-Deficient Mice. Int. J. Mol. Sci. 2022, 23, 8385. [Google Scholar] [CrossRef] [PubMed]
- Steffen, B.T.; Duprez, D.; Szklo, M.; Guan, W.; Tsai, M.Y. Circulating oleic acid levels are related to greater risks of cardiovascular events and all-cause mortality: The Multi-Ethnic Study of Atherosclerosis. J. Clin. Lipidol. 2018, 12, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Qamar, W.; Alqahtani, S.; Ahamad, S.R.; Ali, N.; Altamimi, M.A. Untargeted GC–MS investigation of serum metabolomics of coronary artery disease patients. Saudi J. Biol. Sci. 2020, 27, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, N.; El-Baba, C.; Araji, K.; El-Khoury, R.; Usta, J.; Darwiche, N. The Pentose Phosphate Pathway in Cancer: Regulation and Therapeutic Opportunities. Chemotherapy 2021, 66, 179–191. [Google Scholar] [CrossRef]
- Sun, Q.A.; Runge, M.S.; Madamanchi, N.R. Oxidative stress, NADPH oxidases, and arteries. Hämostaseologie 2016, 36, 77–88. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Grondin, J.A.; Khan, W.I. Tryptophan-derived serotonin-kynurenine balance in immune activation and intestinal inflammation. FASEB J. 2021, 35, e21888. [Google Scholar] [CrossRef]
- Moffett, J.R.; Arun, P.; Puthillathu, N.; Vengilote, R.; Ives, J.A.; Badawy, A.A.; Namboodiri, A.M. Quinolinate as a Marker for Kynurenine Metabolite Formation and the Unresolved Question of NAD+ Synthesis during Inflammation and Infection. Front. Immunol. 2020, 11, 31. [Google Scholar] [CrossRef]
- Hestad, K.; Alexander, J.; Rootwelt, H.; Aaseth, J.O. The Role of Tryptophan Dysmetabolism and Quinolinic Acid in Depressive and Neurodegenerative Diseases. Biomolecules 2022, 12, 998. [Google Scholar] [CrossRef] [PubMed]
- Gáspár, R.; Halmi, D.; Demján, V.; Berkecz, R.; Pipicz, M.; Csont, T. Kynurenine Pathway Metabolites as Potential Clinical Biomarkers in Coronary Artery Disease. Front. Immunol. 2022, 12, 768560. [Google Scholar] [CrossRef] [PubMed]
- Ala, M.; Eftekhar, S.P. The Footprint of Kynurenine Pathway in Cardiovascular Diseases. Int. J. Tryptophan Res. 2022, 15, 11786469221096643. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Variable | Control | HC | HC + Complications | CAD | p-Value |
|---|---|---|---|---|---|
| n | 40 | 35 | 114 | 32 | |
| Gender | 0.475 | ||||
| Male, | 18 (45%), | 16 (45.71%), | 63 (55.26%), | 19 (59.38%), | |
| Female | 22 (55) | 19 (54.29%) | 51 (44.74%) | 13 (40.63%) | |
| Age | 44 (42–46.25) | 45 (42–47) | 51 (45–56) | 54.5 (49–59) | <0.001 |
| PWV m/s | 10.35 (9.4–11.1) | 11.8 (10.3–12.8) | 14.05 (11.9–17.7) | 13.6 (11.2–19.35) | <0.001 |
| BMI (kg/m2) | 28.01 (4.45) | 27.82 (3.89) | 31.01 (5.36) | 31.5 (5.27) | <0.001 |
| Systolic BP (mmHg) | 108 (101–116.25) | 115 (106–124.5) | 133 (120.75–141.75) | 123 (118.25–129.5) | <0.001 |
| Diastolic BP (mmHg) | 71.45 (6.00) | 75.37 (8.45) | 83.87 (12.05) | 76.81 (10.82) | <0.001 |
| Hemoglobin (g/dL) | 13.01 (2.52) | 13.5 (1.87) | 13.96 (1.83) | 13.56 (1.59) | 0.062 |
| RBC (×106/μL) | 4.84 (0.66) | 4.81 (0.53) | 5.06 (0.54) | 4.89 (0.56) | 0.042 |
| WBC (×103/μL) | 6.25 (5.47–7.3) | 6 (5.3–7.62) | 6.8 (6–8.4) | 7.6 (6.25–8.87) | 0.005 |
| Platelets (×103/μL) | 245 (197.25–300.75) | 254.5 (213.25–283.5) | 234 (196–279) | 219.5 (192.25–273.25) | 0.471 |
| Urea (mmol/L) | 4.5 (3.77–5.57) | 4.5 (3.7–5.05) | 4.5 (3.7–5.37) | 5 (4.3–6.17) | 0.157 |
| Creatinine (μmol/L) | 64.5 (55.50–76.25) | 64 (56.5–78.5) | 69.5 (58–80) | 72.5 (62–83.5) | 0.226 |
| ALT (U/L) | 17 (14.75–22.5) | 22 (15–32.5) | 22 (17–31) | 21.5 (17.5–27.75) | 0.024 |
| AST (U/L) | 16 (15–20.25) | 19 (16.5–24) | 18 (15–21) | 18 (15–22) | 0.298 |
| HDL Cholesterol (mmol/L) | 1.40 (1.24–1.57) | 1.28 (1.11–1.49) | 1.18 (1.00–1.40) | 1.185 (0.975–1.51) | 0.009 |
| LDL Cholesterol (mmol/L) | 2.84 (2–3) | 4 (3.88–4.60) | 4 (3.64–4.37) | 2.42 (1.98–3.12) | <0.001 |
| Triglyceride (mmol/L) | 1.04 (0.81–1.32) | 1.9 (1.24–2.54) | 1.93 (1.44–2.58) | 1.5 (0.99–2.10) | <0.001 |
| INR | 1 (1–1.1) | 1 (1–1.1) | 1 (1–1.1) | 1 (1–1.1) | 0.458 |
| TSH (mIU/L) | 1.27 (1.05–1.86) | 1.64 (1.06–2.67) | 1.33 (0.90–1.94) | 1.44 (0.90–2.30) | 0.268 |
| HbA1c % | 5.4 (5.1–5.5) | 5.5 (5.15–5.8) | 6.5 (5.8–8.07) | 6.3 (5.7–7.17) | <0.001 |
| CRP (mg/L) | 5 (5–6) | 5 (5–7.5) | 5 (5–8) | 5 (5–9) | 0.721 |
| Metabolites | Sub-Pathway | Super-Pathway | Estimate | SE | p-Value | FDR |
|---|---|---|---|---|---|---|
| Mannitol/Sorbitol | Fructose, Mannose, and Galactose Metabolism | Carbohydrate | 0.447 | 0.097 | <0.001 | 0.005 |
| Glucose | Glycolysis, Gluconeogenesis, and Pyruvate Metabolism | Carbohydrate | 0.156 | 0.041 | <0.001 | 0.062 |
| Pregnenediol Sulfate (C21H34O5S) | Pregnenolone Steroids | Lipid | −0.314 | 0.084 | <0.001 | 0.062 |
| Mannose | Fructose, Mannose, and Galactose Metabolism | Carbohydrate | 0.171 | 0.048 | <0.001 | 0.078 |
| Oleoylcarnitine (C18:1) | Fatty Acid Metabolism (Acyl Carnitine, Monounsaturated) | Lipid | −0.150 | 0.042 | <0.001 | 0.078 |
| Ribitol | Pentose Metabolism | Carbohydrate | 0.137 | 0.040 | <0.001 | 0.092 |
| Quinolinate | Nicotinate and Nicotinamide Metabolism | Cofactors and Vitamins | −0.150 | 0.044 | <0.001 | 0.092 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anlar, G.G.; Anwardeen, N.; Al Ashmar, S.; Pedersen, S.; Elrayess, M.A.; Zeidan, A. Metabolomics Profiling of Stages of Coronary Artery Disease Progression. Metabolites 2024, 14, 292. https://doi.org/10.3390/metabo14060292
Anlar GG, Anwardeen N, Al Ashmar S, Pedersen S, Elrayess MA, Zeidan A. Metabolomics Profiling of Stages of Coronary Artery Disease Progression. Metabolites. 2024; 14(6):292. https://doi.org/10.3390/metabo14060292
Chicago/Turabian StyleAnlar, Gulsen Guliz, Najeha Anwardeen, Sarah Al Ashmar, Shona Pedersen, Mohamed A. Elrayess, and Asad Zeidan. 2024. "Metabolomics Profiling of Stages of Coronary Artery Disease Progression" Metabolites 14, no. 6: 292. https://doi.org/10.3390/metabo14060292
APA StyleAnlar, G. G., Anwardeen, N., Al Ashmar, S., Pedersen, S., Elrayess, M. A., & Zeidan, A. (2024). Metabolomics Profiling of Stages of Coronary Artery Disease Progression. Metabolites, 14(6), 292. https://doi.org/10.3390/metabo14060292