

Mass Spectrometric Analysis of Purine Intermediary Metabolism Indicates Cyanide Induces Purine Catabolism in Rabbits


Abstract
1. Introduction
2. Experimental Procedures
2.1. Rabbits
2.2. Diffuse Optical Spectroscopy (DOS)
2.3. Oxypurinol Administration
2.4. Glyoxylate and Hexachloroplaninate Administration
2.5. Targeted Metabolomics
2.6. Pharmacokinetics of Oxypurinol
2.7. Zebrafish
2.8. Statistics
3. Results
3.1. Plasma Levels of Purine Metabolites Increase in Rabbits Exposed to a Lethal Dose of Cyanide
3.2. Pharmacological Inhibition of Purine Catabolism Confers Protection against Cyanide Poisoning in Zebrafish
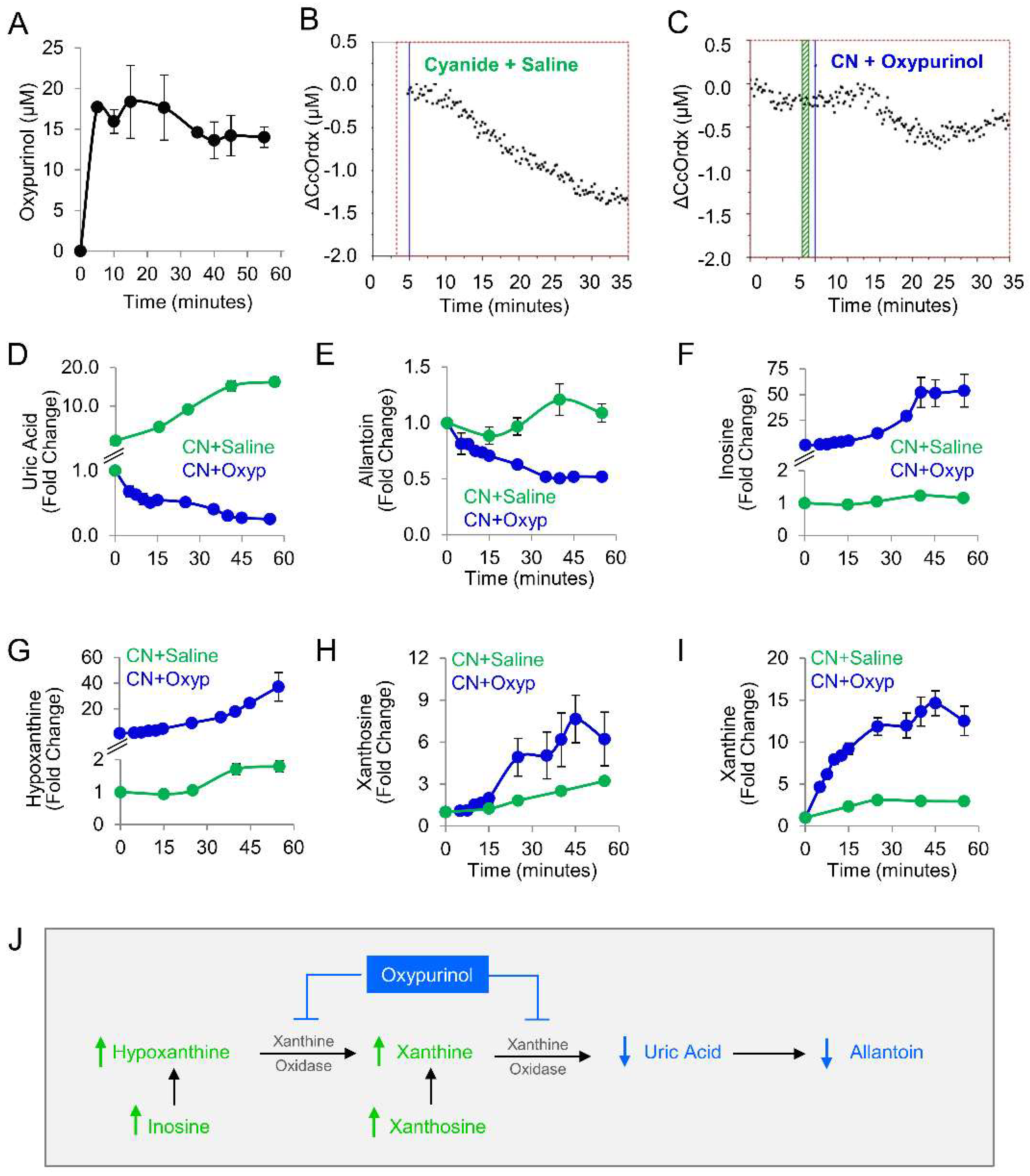
3.3. Oxypurinol Abrogates the Deleterious Effects of Cyanide on Cytochrome c Oxidase in Rabbits Exposed to a Lethal Dose of Cyanide
3.4. Uric Acid Correlates with Known Biomarkers of Cyanide Poisoning
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yadav, M.; Pulletikurti, S.; Yerabolu, J.R.; Krishnamurthy, R. Cyanide as a primordial reductant enables a protometabolic reductive glyoxylate pathway. Nat. Chem. 2022, 14, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Szabo, C. The two faces of cyanide: An environmental toxin and a potential novel mammalian gasotransmitter. FEBS J. 2022, 289, 2481–2515. [Google Scholar] [CrossRef] [PubMed]
- Isom, G.E.; Way, J.L. Effects of oxygen on the antagonism of cyanide intoxication: Cytochrome oxidase, in vitro. Toxicol. Appl. Pharmacol. 1984, 74, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Leavesley, H.B.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. Interaction of Cyanide and Nitric Oxide with Cytochrome c Oxidase: Implications for Acute Cyanide Toxicity. Toxicol. Sci. 2008, 101, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, P.; Soulimane, T. The mixed valence state of the oxidase binuclear centre: How Thermus thermophilus cytochrome ba3 differs from classical aa3 in the aerobic steady state and when inhibited by cyanide. Biochim. Biophys. Acta (BBA) Bioenerg. 2004, 1655, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Robinson, N.C. Kinetics and mechanism for the binding of HCN to cytochrome c oxidase. Biochemistry 1995, 34, 10009–10018. [Google Scholar] [CrossRef]
- Gawron, O. Chapter 14—On the reaction of cyanide with cystine and cystine peptides. In The Chemistry of Organic Sulfur Compounds; Kharasch, N., Meyers, C.Y., Eds.; Pergamon: Bergama, Turkey, 1966; pp. 351–365. [Google Scholar]
- García, I.; Arenas-Alfonseca, L.; Moreno, I.; Gotor, C.; Romero, L.C. HCN Regulates Cellular Processes through Posttranslational Modification of Proteins by S-cyanylation. Plant Physiol. 2019, 179, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, O.A.; Bortey-Sam, N.; Mahon, S.B.; Brenner, M.; Rockwood, G.A.; Logue, B.A. Metabolism of Cyanide by Glutathione to Produce the Novel Cyanide Metabolite 2-Aminothiazoline-4-oxoaminoethanoic Acid. Chem. Res. Toxicol. 2019, 32, 718–726. [Google Scholar] [CrossRef]
- Fasco, M.J.; Hauer, C.R.; Stack, R.F.; O’Hehir, C.; Barr, J.R.; Eadon, G.A. Cyanide Adducts with Human Plasma Proteins: Albumin as a Potential Exposure Surrogate. Chem. Res. Toxicol. 2007, 20, 677–684. [Google Scholar] [CrossRef]
- Moore, S.J.; Norris, J.C.; Ho, I.K.; Hume, A.S. The efficacy of α-ketoglutaric acid in the antagonism of cyanide intoxication. Toxicol. Appl. Pharmacol. 1986, 82, 40–44. [Google Scholar] [CrossRef]
- Schwartz, C.; Morgan, R.L.; Way, L.M.; Way, J.L. Antagonism of cyanide intoxication with sodium pyruvate. Toxicol. Appl. Pharmacol. 1979, 50, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Isom, G.E.; Borowitz, J.L. Biochemical mechanisms of cyanide toxicity. In Toxicology of Cyanides and Cyanogens; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 70–81. [Google Scholar]
- Nath, A.K.; Roberts, L.D.; Liu, Y.; Mahon, S.B.; Kim, S.; Ryu, J.H.; Werdich, A.; Januzzi, J.L.; Boss, G.R.; Rockwood, G.A.; et al. Chemical and metabolomic screens identify novel bi-omarkers and antidotes for cyanide exposure. FASEB J. 2013, 27, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Sips, P.Y.; Shi, X.; Musso, G.; Nath, A.K.; Zhao, Y.; Nielson, J.; Morningstar, J.; Kelly, A.E.; Mikell, B.; Buys, E.; et al. Identification of specific metabolic pathways as druggable targets regulating the sensitivity to cyanide poisoning. PLoS ONE 2018, 13, e0193889. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Purine Metabolism. Available online: https://pubchem.ncbi.nlm.nih.gov/pathway/PathBank:SMP0000050 (accessed on 7 May 2024).
- Massey, V.; Edmondson, D. On the Mechanism of Inactivation of Xanthine Oxidase by Cyanide. J. Biol. Chem. 1970, 245, 6595–6598. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.; Johnson, J.; Rajagopalan, K. Mechanisms of inactivation of molybdoenzymes by cyanide. J. Biol. Chem. 1980, 255, 2694–2699. [Google Scholar] [CrossRef] [PubMed]
- ATSDR. Toxicological Profile for Cyanide; United States Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 1997. [Google Scholar]
- Malthouse, J.P.; Bray, R.C. The nature of the sulphur atom liberated from xanthine oxidase by cyanide. Evidence from e.p.r. spectroscopy after 35S substitution. Biochem. J. 1980, 191, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Lee, J.; Mohammad, O.; Mahon, S.B.; Sharma, V.S.; Kim, J.; Mukai, D.; Blackledge, W.; Boss, G.R.; Goodman, S.; et al. Comparison of cobinamide to hydroxocobalamin in reversing cyanide physiologic effects in rabbits using diffuse optical spectroscopy monitoring. J. Biomed. Opt. 2010, 15, 017001. [Google Scholar] [CrossRef]
- Lee, J.; Armstrong, J.; Kreuter, K.; Tromberg, B.J.; Brenner, M. Non-invasive in vivo diffuse optical spectroscopy monitoring of cyanide poisoning in a rabbit model. Physiol. Meas. 2007, 28, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.K.; Shi, X.; Harrison, D.L.; Morningstar, J.E.; Mahon, S.; Chan, A.; Sips, P.; Lee, J.; MacRae, C.A.; Boss, G.R.; et al. Cisplatin Analogs Confer Protection against Cyanide Poisoning. Cell Chem. Biol. 2017, 24, 565–575.e4. [Google Scholar] [CrossRef]
- Nielson, J.R.; Nath, A.K.; Doane, K.P.; Shi, X.; Lee, J.; Tippetts, E.G.; Saha, K.; Morningstar, J.; Hicks, K.G.; Chan, A.; et al. Glyoxylate protects against cyanide toxicity through metabolic modulation. Sci. Rep. 2022, 12, 1–16. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.G.; Mahon, S.B.; Mukai, D.; Yoon, D.; Boss, G.R.; Patterson, S.E.; Rockwood, G.; Isom, G.; Brenner, M. Noninvasive optical cytochrome c oxidase redox state measurements using diffuse optical spectroscopy. J. Biomed. Opt. 2014, 19, 055001. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Rockwood, G.; Logue, B.; Manandhar, E.; Petrikovics, I.; Han, C.; Bebarta, V.; Mahon, S.B.; Burney, T.; Brenner, M. Monitoring Dose Response of Cyanide Antidote Dimethyl Trisulfide in Rabbits Using Diffuse Optical Spectroscopy. J. Med. Toxicol. 2018, 14, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.R.; Moody, A.J. Cytochrome c oxidase. In Bioenergetics; Gräber, P., Milazzo, G., Walz, D., Eds.; Birkhäuser: Basel, Switzerland, 1997; pp. 418–456. [Google Scholar]
- Morningstar, J.; Lee, J.; Hendry-Hofer, T.; Witeof, A.; Lyle, L.T.; Knipp, G.; MacRae, C.A.; Boss, G.R.; Peterson, R.T.; Davisson, V.J.; et al. Intramuscular administration of hexachloroplatinate reverses cyanide-induced metabolic derangements and counteracts severe cyanide poisoning. FASEB Bioadv. 2019, 1, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Bebarta, V.S.; Shi, X.; Zheng, S.; Hendry-Hofer, T.B.; Severance, C.C.; Behymer, M.M.; Boss, G.R.; Mahon, S.; Brenner, M.; Knipp, G.T.; et al. Intramuscular administration of glyoxylate rescues swine from lethal cyanide poisoning and ameliorates the biochemical sequalae of cyanide intoxication. Toxicol. Sci. 2023, 191, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Makarewicz, W. Response of purine metabolism to hypoxia and ischemia. Adv. Exp. Med. Biol. 1998, 431, 351–357. [Google Scholar] [PubMed]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Harkness, R. Hypoxanthine, xanthine and uridine in body fluids, indicators of ATP depletion. J. Chromatogr. B Biomed. Sci. Appl. 1988, 429, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Beckman, J.S.; Beckman, T.K.; Wheat, J.K.; Cash, T.G.; Freeman, B.A.; Parks, D.A. Circulating xanthine oxidase: Potential mediator of ischemic injury. Am. J. Physiol. Liver Physiol. 1990, 258, G564–G570. [Google Scholar] [CrossRef]
- Kayyali, U.S.; Donaldson, C.; Huang, H.; Abdelnour, R.; Hassoun, P.M. Phosphorylation of xanthine dehydrogenase/oxidase in hypoxia. J. Biol. Chem. 2001, 276, 14359–14365. [Google Scholar] [CrossRef]
- Amaya, Y.; Yamazaki, K.; Sato, M.; Noda, K.; Nishino, T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J. Biol. Chem. 1990, 265, 14170–14175. [Google Scholar] [CrossRef]
- Hille, R.; Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995, 9, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Fukushima, T.; Usami, Y.; Hirano, K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem. J. 1993, 289 Pt 2, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.; Estevez, A.; Chumley, P.; Aslan, M.; Marklund, S.; Parks, D.A.; Freeman, B.A. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. J. Biol. Chem. 1999, 274, 4985–4994. [Google Scholar] [CrossRef]
- Johnson, J.D.; Conroy, W.G.; Burris, K.D.; Isom, G.E. Peroxidation of brain lipids following cyanide intoxication in mice. Toxicology 1987, 46, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Belani, K.; Hottinger, D.; Beebe, D.; Kozhimannil, T.; Prielipp, R. Sodium nitroprusside in 2014: A clinical concepts review. J. Anaesthesiol. Clin. Pharmacol. 2014, 30, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.M.; Splawinski, Z.; Bott, R.C.; Logan, M.B. Combustion products generated in simulated industrial fires. J. Occup. Environ. Hyg. 2021, 18, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Fent, K.W.; Evans, D.E.; Babik, K.; Striley, C.; Bertke, S.; Kerber, S.; Smith, D.; Horn, G.P. Airborne contaminants during controlled residential fires. J. Occup. Environ. Hyg. 2018, 15, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Mehrotra, M.; Kumar, P.; Gogia, A.R.; Prasad, A.; Fisher, J.A. Smoke Inhalation Injury: Etiopathogenesis, Diagnosis, and Management. Indian J. Crit. Care Med. 2018, 22, 180–188. [Google Scholar] [CrossRef]
- Baud, F.J.; Barriot, P.; Toffis, V.; Riou, B.; Vicaut, E.; Lecarpentier, Y.; Bourdon, R.; Astier, A.; Bismuth, C. Elevated Blood Cyanide Concentrations in Victims of Smoke Inhalation. N. Engl. J. Med. 1991, 325, 1761–1766. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Saline (n = 5) | Cyanide (n = 15) | p-Value | q-Value |
|---|---|---|---|---|
| Glucose | 1.07 ± 0.05 | 0.82 ± 0.10 | 0.0249 | 0.002802 |
| Pyruvic Acid | 1.22 ± 0.22 | 8.43 ± 1.46 | 0.0001 | 0.000028 |
| Lactic Acid | 1.16 ± 0.17 | 3.61 ± 0.95 | 0.0081 | 0.001030 |
| Oxaloacetic Acid | 0.89 ± 0.03 | 3.31 ± 0.35 | <0.0001 | 0.000025 |
| Aconitic Acid | 0.89 ± 0.05 | 1.70 ± 0.13 | 0.0001 | 0.000028 |
| Alpha-Ketoglutaric Acid | 0.78 ± 0.05 | 3.68 ± 0.55 | <0.0001 | 0.000025 |
| Succinic Acid | 0.97 ± 0.14 | 24.04 ± 4.37 | <0.0001 | 0.000025 |
| Fumaric Acid | 1.14 ± 0.14 | 15.02 ± 2.87 | <0.0001 | 0.000025 |
| Malic Acid | 1.07 ± 0.13 | 4.55 ± 0.61 | 0.0001 | 0.000028 |
| Metabolite | Saline (n = 5) | Cyanide (n = 12) | p-Value | q-Value |
|---|---|---|---|---|
| Uric Acid | 1.28 ± 0.19 | 16.28 ± 1.27 | 0.0006 | 0.00033 |
| Xanthosine | 0.69 ± 0.07 | 3.23 ± 0.19 | 0.0006 | 0.00033 |
| Xanthine | 0.71 ± 0.08 | 2.95 ± 0.16 | 0.0006 | 0.00033 |
| Hypoxanthine | 0.37 ± 0.12 | 1.79 ± 0.17 | 0.0013 | 0.00053 |
| Inosine | 0.41 ± 0.10 | 1.08 ± 0.08 | 0.0046 | 0.00157 |
| AMP | 1.27 ± 0.50 | 3.74 ± 0.98 | 0.0992 | 0.02863 |
| Adenosine | 0.68 ± 0.08 | 1.72 ± 0.47 | 0.2544 | 0.06423 |
| Allantoin | 0.73 ± 0.04 | 1.15 ± 0.03 | 0.0006 | 0.00033 |
| Metabolite | Cyanide (n = 12) | Cyanide + Oxypurinol (n = 4) | p-Value | q-Value |
|---|---|---|---|---|
| Uric Acid | 16.28 ± 1.27 | 0.25 ± 0.01 | 0.0006 | 0.00040 |
| Xanthosine | 3.23 ± 0.19 | 6.21 ± 1.91 | 0.0992 | 0.01670 |
| Xanthine | 2.95 ± 0.16 | 12.52 ± 1.74 | 0.0006 | 0.00168 |
| Hypoxanthine | 1.79 ± 0.17 | 37.29 ± 11.19 | 0.0006 | 0.00168 |
| Inosine | 1.08 ± 0.08 | 53.71 ± 15.92 | 0.0006 | 0.00168 |
| Allantoin | 1.15 ± 0.03 | 0.51 ± 0.01 | 0.0019 | 0.00040 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morningstar, J.; Lee, J.; Mahon, S.; Brenner, M.; Nath, A.K. Mass Spectrometric Analysis of Purine Intermediary Metabolism Indicates Cyanide Induces Purine Catabolism in Rabbits. Metabolites 2024, 14, 279. https://doi.org/10.3390/metabo14050279
Morningstar J, Lee J, Mahon S, Brenner M, Nath AK. Mass Spectrometric Analysis of Purine Intermediary Metabolism Indicates Cyanide Induces Purine Catabolism in Rabbits. Metabolites. 2024; 14(5):279. https://doi.org/10.3390/metabo14050279
Chicago/Turabian StyleMorningstar, Jordan, Jangwoen Lee, Sari Mahon, Matthew Brenner, and Anjali K. Nath. 2024. "Mass Spectrometric Analysis of Purine Intermediary Metabolism Indicates Cyanide Induces Purine Catabolism in Rabbits" Metabolites 14, no. 5: 279. https://doi.org/10.3390/metabo14050279
APA StyleMorningstar, J., Lee, J., Mahon, S., Brenner, M., & Nath, A. K. (2024). Mass Spectrometric Analysis of Purine Intermediary Metabolism Indicates Cyanide Induces Purine Catabolism in Rabbits. Metabolites, 14(5), 279. https://doi.org/10.3390/metabo14050279