Isobaric Tags for Relative and Absolute Quantitation-Based Proteomics Analysis Revealed Proteins Involved in Drought Response during the Germination Stage in Faba Bean

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Protein Extraction and Trypsin Digestion
2.3. iTRAQ Labeling and LC-MS/MS Analysis
2.4. Database Search
2.5. Gene Ontology Analysis Enrichment
2.6. Pathway Analysis Enrichment
2.7. Protein–Protein Interaction Network
2.8. RNA Extraction and qRT-PCR
2.9. Silencing, Ectopic Expression of Targets, and RT-PCR
2.10. Physiological Parameter Measurements and Statistical Analysis
3. Result
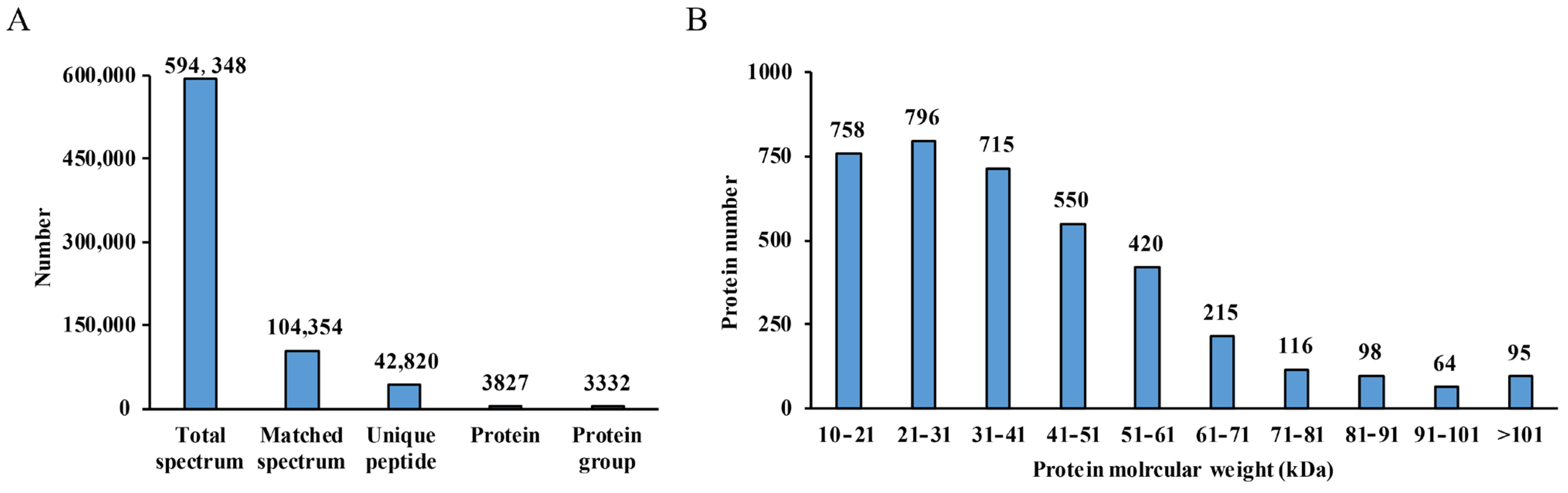
3.1. Quantitative Proteomic Analysis
3.2. Identification of DEPs
3.3. Functional Annotation of DEPs
3.4. Protein–Protein Interaction Analysis
3.5. Quantitative Real-Time Polymerase Chain Reaction Assay
3.6. Function Validation of the DEPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amine-Khodja, I.R.; Boscari, A.; Riah, N.; Kechid, M.; Maougal, R.T.; Belbekri, N.; Djekoun, A. Impact of Two Strains of Rhizobium leguminosarum on the Adaptation to Terminal Water Deficit of Two Cultivars Vicia faba. Plants 2022, 11, 515. [Google Scholar] [CrossRef]
- Dietz, K.J.; Zörb, C.; Geilfus, C.M. Drought and crop yield. Plant Biol. 2021, 23, 881–893. [Google Scholar] [CrossRef]
- Marthandan, V.; Geetha, R.; Kumutha, K.; Renganathan, V.G.; Karthikeyan, A.; Ramalingam, J. Seed priming: A feasible strategy to enhance drought tolerance in crop plants. Int. J. Mol. Sci. 2020, 21, 8258. [Google Scholar] [CrossRef]
- Thabet, S.G.; Moursi, Y.S.; Karam, M.A.; Graner, A.; Alqudah, A.M. Genetic basis of drought tolerance during seed germination in barley. PLoS ONE 2018, 13, e0206682. [Google Scholar] [CrossRef]
- Li, P.; Zhang, Y.; Wu, X.; Liu, Y. Drought stress impact on leaf proteome variations of faba bean (Vicia faba L.) in the Qinghai-Tibet Plateau of China. 3 Biotech 2018, 8, 110. [Google Scholar] [CrossRef]
- Parvin, S.; Uddin, S.; Tausz-Posch, S.; Fitzgerald, G.; Armstrong, R.; Tausz, M. Elevated CO2 improves yield and N2 fxation but not grain N concentration of faba bean (Vicia faba L.) subjected to terminal drought. Environ. Exp. Bot. 2019, 165, 161–173. [Google Scholar] [CrossRef]
- Jayakodi, M.; Golicz, A.A.; Kreplak, J.; Fechete, L.I.; Angra, D.; Bednář, P.; Bornhofen, E.; Zhang, H.; Boussageon, R.; Kaur, S.; et al. The giant diploid faba genome unlocks variation in a global protein crop. Nature 2023, 615, 652–659. [Google Scholar] [CrossRef]
- Ammar, M.H.; Khan, A.M.; Migdadi, H.M.; Abdelkhalek, S.M.; Alghamdi, S.S. Faba bean drought responsive gene identification and validation. Saudi J. Biol. Sci. 2017, 24, 80–89. [Google Scholar] [CrossRef]
- Alghamdi, S.S.; Khan, M.A.; Ammar, M.H.; Sun, Q.; Huang, L.; Migdadi, H.M.; El-Harty, E.H.; Al-Faifi, S.A. Characterization of drought stress-responsive root transcriptome of faba bean (Vicia faba L.) using RNA sequencing. 3 Biotech 2018, 8, 502. [Google Scholar] [CrossRef]
- Wu, X.; Fan, Y.; Li, L.; Liu, Y. The influence of soil drought stress on the leaf transcriptome of faba bean (Vicia faba L.) in the Qinghai-Tibet Plateau. 3 Biotech 2020, 10, 381. [Google Scholar] [CrossRef]
- Khazaei, H.; O’Sullivan, D.M.; Sillanpää, M.J.; Stoddard, F.L. Use of synteny to identify candidate genes underlying QTL controlling stomatal traits in faba bean (Vicia faba L.). Theor. Appl. Genet. 2014, 127, 2371–2385. [Google Scholar] [CrossRef]
- Liu, G.; Chen, J.; Wang, X. VfCPK1, a gene encoding calcium-dependent protein kinase from Vicia faba, is induced by drought and abscisic acid. Plant Cell Environ. 2006, 29, 2091–2099. [Google Scholar] [CrossRef]
- Qie, L.; Jia, G.; Zhang, W.; Schnable, J.; Shang, Z.; Li, W.; Liu, B.; Li, M.; Chai, Y.; Zhi, H.; et al. Mapping of Quantitative trait locus (QTLs) that contribute to germination and early seedling drought tolerance in the interspecific cross Setaria italica × Setaria viridis. PLoS ONE 2014, 9, e101868. [Google Scholar] [CrossRef]
- Gad, M.; Chao, H.; Li, H.; Zhao, W.; Lu, G.; Li, M. QTL mapping for seed germination response to drought stress in Brassica napus. Front. Plant Sci. 2021, 11, 629970. [Google Scholar] [CrossRef]
- Liu, S.; Zenda, T.; Dong, A.; Yang, Y.; Liu, X.; Wang, Y.; Li, J.; Tao, Y.; Duan, H. Comparative proteomic and morpho-physiological analyses of maize wild-type Vp16 and mutant vp16 germinating seed responses to PEG-induced drought stress. Int. J. Mol. Sci. 2019, 20, 5586. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, S.; Guo, X.; Jia, J.; Yang, W.; Liu, Z.; Chen, S.; Li, X.; Qi, D.; Liu, G.; et al. A MYB-related transcription factor from sheepgrass, LcMYB2, promotes seed germination and root growth under drought stress. BMC Plant Biol. 2019, 19, 564. [Google Scholar] [CrossRef]
- Yu, A.; Zhao, J.; Wang, Z.; Cheng, K.; Zhang, P.; Tian, G.; Liu, X.; Guo, E.; Du, Y.; Wang, Y. Transcriptome and metabolite analysis reveal the drought tolerance of foxtail millet significantly correlated with phenylpropanoids-related pathways during germination process under PEG stress. BMC Plant Biol. 2020, 20, 274. [Google Scholar] [CrossRef]
- Zhu, Z.; Chen, H.; Xie, K.; Liu, C.; Li, L.; Liu, L.; Han, X.; Jiao, C.; Wan, Z.; Sha, A. Characterization of drought-responsive transcriptome during seed germination in adzuki bean (Vigna angularis L.) by PacBio SMRT and Illumina Sequencing. Front. Genet. 2020, 11, 996. [Google Scholar] [CrossRef]
- Han, X.; Yang, F.; Zhao, Y.; Chen, H.; Wan, Z.; Li, L.; Sun, L.; Liu, L.; Jiao, C.; Liu, C.; et al. iTRAQ based protein profile analysis revealed key proteins involved in regulation of drought-tolerance during seed germination in Adzuki bean. Sci. Rep. 2021, 11, 23725. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, C.; Chen, H.; Li, L.; Sun, L.; Han, X.; Jiao, C.; Sha, A. Analysis of ASPAT gene family based on drought-stressed transcriptome sequencing in Vicia faba L. Acta Agron. Sin. 2023, 49, 1871–1881. [Google Scholar]
- Riyazuddin, R.; Nisha, N.; Singh, K.; Verma, R.; Gupta, R. Involvement of dehydrin proteins in mitigating the negative effects of drought stress in plants. Plant Cell Rep. 2022, 41, 519–533. [Google Scholar] [CrossRef]
- Cho, S.K.; Kim, J.E.; Park, J.A.; Eom, T.J.; Kim, W.T. Constitutive expression of abiotic stress-inducible hot pepper CaXTH3, which encodes a xyloglucan endotransglucosylase/hydrolase homolog, improves drought and salt tolerance in transgenic Arabidopsis plants. FEBS Lett. 2006, 580, 3136–3144. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamaguchi-Shinozaki, K. ABA signaling in stress-response and seed development. Plant Cell Rep. 2013, 32, 959–970. [Google Scholar] [CrossRef]
- Shiraku, M.L.; Magwanga, R.O.; Zhang, Y.; Hou, Y.; Kirungu, J.N.; Mehari, T.G.; Xu, Y.; Wang, Y.; Wang, K.; Cai, X.; et al. Late embryogenesis abundant gene LEA3 (Gh_A08G0694) enhances drought and salt stress tolerance in cotton. Int. J. Biol. Macromol. 2022, 207, 700–714. [Google Scholar] [CrossRef]
- Tamang, T.M.; Sprague, S.A.; Kakeshpour, T.; Liu, S.; White, F.F.; Park, S. Ectopic Expression of a Heterologous Glutaredoxin Enhances Drought Tolerance and Grain Yield in Field Grown Maize. Int. J. Mol. Sci. 2021, 22, 5331. [Google Scholar] [CrossRef]
- Yang, F.; Chen, H.; Liu, C.; Li, L.; Liu, L.; Han, X.; Wan, Z.; Sha, A. Transcriptome profile analysis of two Vicia faba cultivars with contrasting salinity tolerance during seed germination. Sci. Rep. 2020, 10, 7250. [Google Scholar] [CrossRef]
- Ibrahim, E.A. Seed priming to alleviate salinity stress in germinating seeds. J. Plant Physiol. 2016, 192, 38–46. [Google Scholar] [CrossRef]
- Petibon, C.; Malik Ghulam, M.; Catala, M.; Abou Elela, S. Regulation of ribosomal protein genes: An ordered anarchy. Wiley Interdiscip. Rev. RNA 2021, 12, e1632. [Google Scholar] [CrossRef]
- Moin, M.; Bakshi, A.; Madhav, M.S.; Kirti, P.B. Expression Profiling of Ribosomal Protein Gene Family in Dehydration Stress Responses and Characterization of Transgenic Rice Plants Overexpressing RPL23A for Water-Use Efficiency and Tolerance to Drought and Salt Stresses. Front. Chem. 2017, 5, 97. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, B.; Jiang, C.; Ming, F. RceIF5A, encoding an eukaryotic translation initiation factor 5A in Rosa chinensis, can enhance thermotolerance, oxidative and osmotic stress resistance of Arabidopsis thaliana. Plant Mol. Biol. 2011, 75, 167–178. [Google Scholar] [CrossRef]
- Fíla, J.; Klodová, B.; Potěšil, D.; Juříček, M.; Šesták, P.; Zdráhal, Z.; Honys, D. The beta Subunit of Nascent Polypeptide Associated Complex Plays A Role in Flowers and Siliques Development of Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 2065. [Google Scholar] [CrossRef]
- Li, H.; Xu, H.; Graham, D.E.; White, R.H. The Methanococcus jannaschii dCTP deaminase is a bifunctional deaminase and diphosphatase. J. Biol. Chem. 2003, 278, 11100–11106. [Google Scholar] [CrossRef]
- Liu, B.; Cyr, R.J.; Palevitz, B.A. A kinesin-like protein, KatAp, in the cells of Arabidopsis and other plants. Plant Cell 1996, 8, 119–132. [Google Scholar]
- Gao, J.; Zhang, S.; He, W.D.; Shao, X.H.; Li, C.Y.; Wei, Y.R.; Deng, G.M.; Kuang, R.B.; Hu, C.H.; Yi, G.J.; et al. Comparative Phosphoproteomics Reveals an Important Role of MKK2 in Banana (Musa spp.) Cold Signal Network. Sci. Rep. 2017, 7, 40852. [Google Scholar] [CrossRef]
- Islam, M.M.; Sandhu, J.; Walia, H.; Saha, R. Transcriptomic data-driven discovery of global regulatory features of rice seeds developing under heat stress. Comput. Struct. Biotechnol. J. 2020, 18, 2556–2567. [Google Scholar] [CrossRef]
- Rizvi, I.; Choudhury, N.R.; Tuteja, N. Arabidopsis thaliana MCM3 single subunit of MCM2-7 complex functions as 3′ to 5′ DNA helicase. Protoplasma 2016, 253, 467–475. [Google Scholar] [CrossRef]
- Li, S.; He, X.; Gao, Y.; Zhou, C.; Chiang, V.L.; Li, W. Histone Acetylation Changes in Plant Response to Drought Stress. Genes 2021, 12, 1409. [Google Scholar] [CrossRef]
- Hou, J.; Ren, R.; Xiao, H.; Chen, Z.; Yu, J.; Zhang, H.; Shi, Q.; Hou, H.; He, S.; Li, L. Characteristic and evolution of HAT and HDAC genes in Gramineae genomes and their expression analysis under diverse stress in Oryza sativa. Planta 2021, 253, 72. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Yang, F.; Li, L.; Han, X.; Chen, H.; Sha, A.; Jiao, C. Isobaric Tags for Relative and Absolute Quantitation-Based Proteomics Analysis Revealed Proteins Involved in Drought Response during the Germination Stage in Faba Bean. Metabolites 2024, 14, 175. https://doi.org/10.3390/metabo14030175
Liu C, Yang F, Li L, Han X, Chen H, Sha A, Jiao C. Isobaric Tags for Relative and Absolute Quantitation-Based Proteomics Analysis Revealed Proteins Involved in Drought Response during the Germination Stage in Faba Bean. Metabolites. 2024; 14(3):175. https://doi.org/10.3390/metabo14030175
Chicago/Turabian StyleLiu, Changyan, Fangwen Yang, Li Li, Xuesong Han, Hongwei Chen, Aihua Sha, and Chunhai Jiao. 2024. "Isobaric Tags for Relative and Absolute Quantitation-Based Proteomics Analysis Revealed Proteins Involved in Drought Response during the Germination Stage in Faba Bean" Metabolites 14, no. 3: 175. https://doi.org/10.3390/metabo14030175
APA StyleLiu, C., Yang, F., Li, L., Han, X., Chen, H., Sha, A., & Jiao, C. (2024). Isobaric Tags for Relative and Absolute Quantitation-Based Proteomics Analysis Revealed Proteins Involved in Drought Response during the Germination Stage in Faba Bean. Metabolites, 14(3), 175. https://doi.org/10.3390/metabo14030175