
Machine Learning to Predict Enzyme–Substrate Interactions in Elucidation of Synthesis Pathways: A Review

 , , ,
, , , 
Abstract
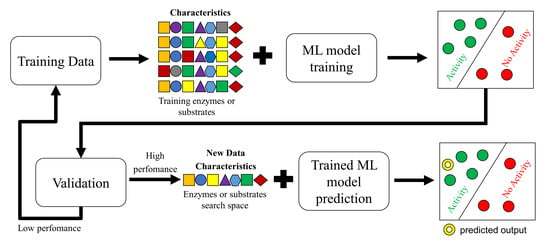
1. Introduction
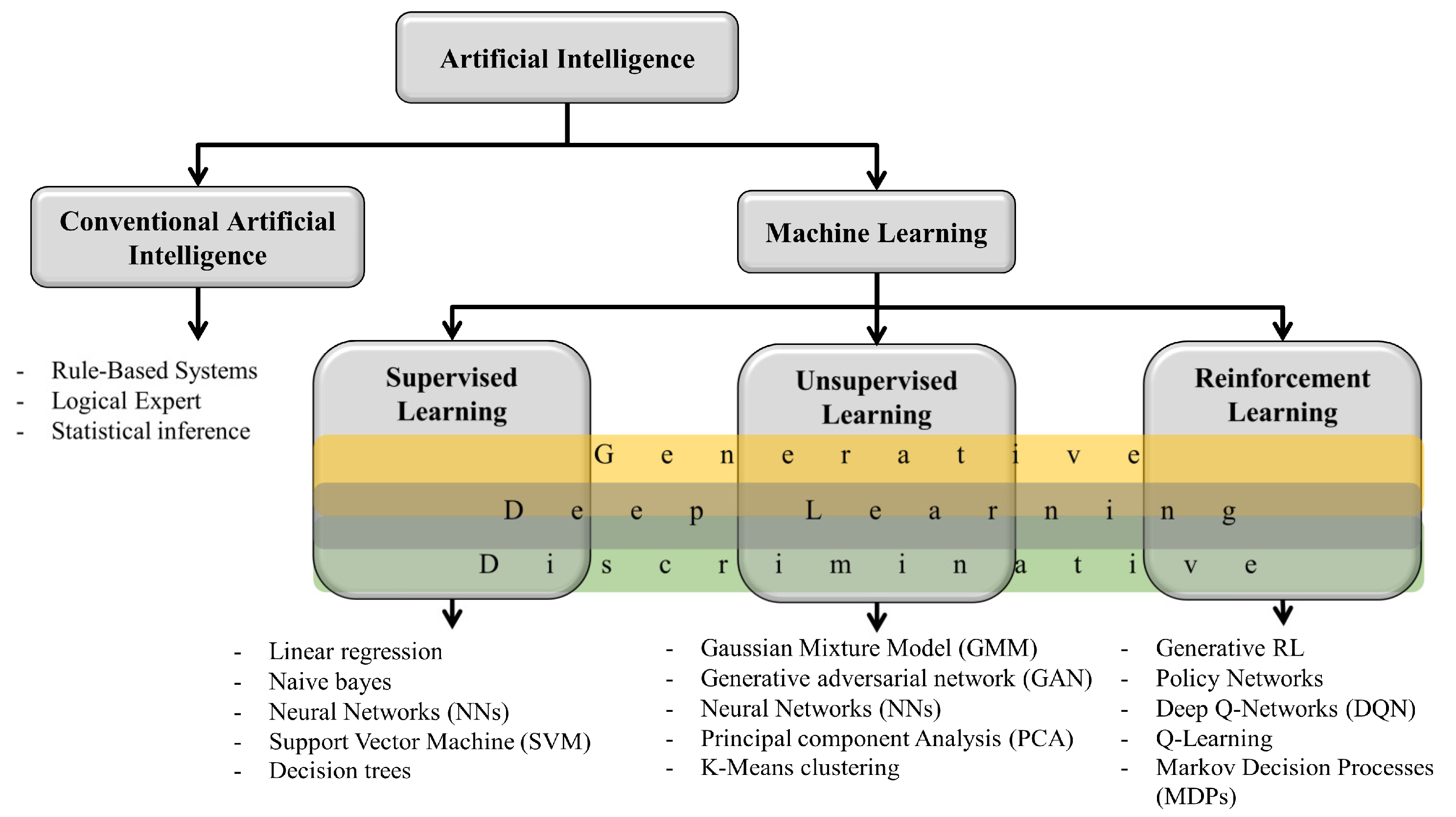
2. Fundamentals of Artificial Intelligence
3. Training Data—Data Set
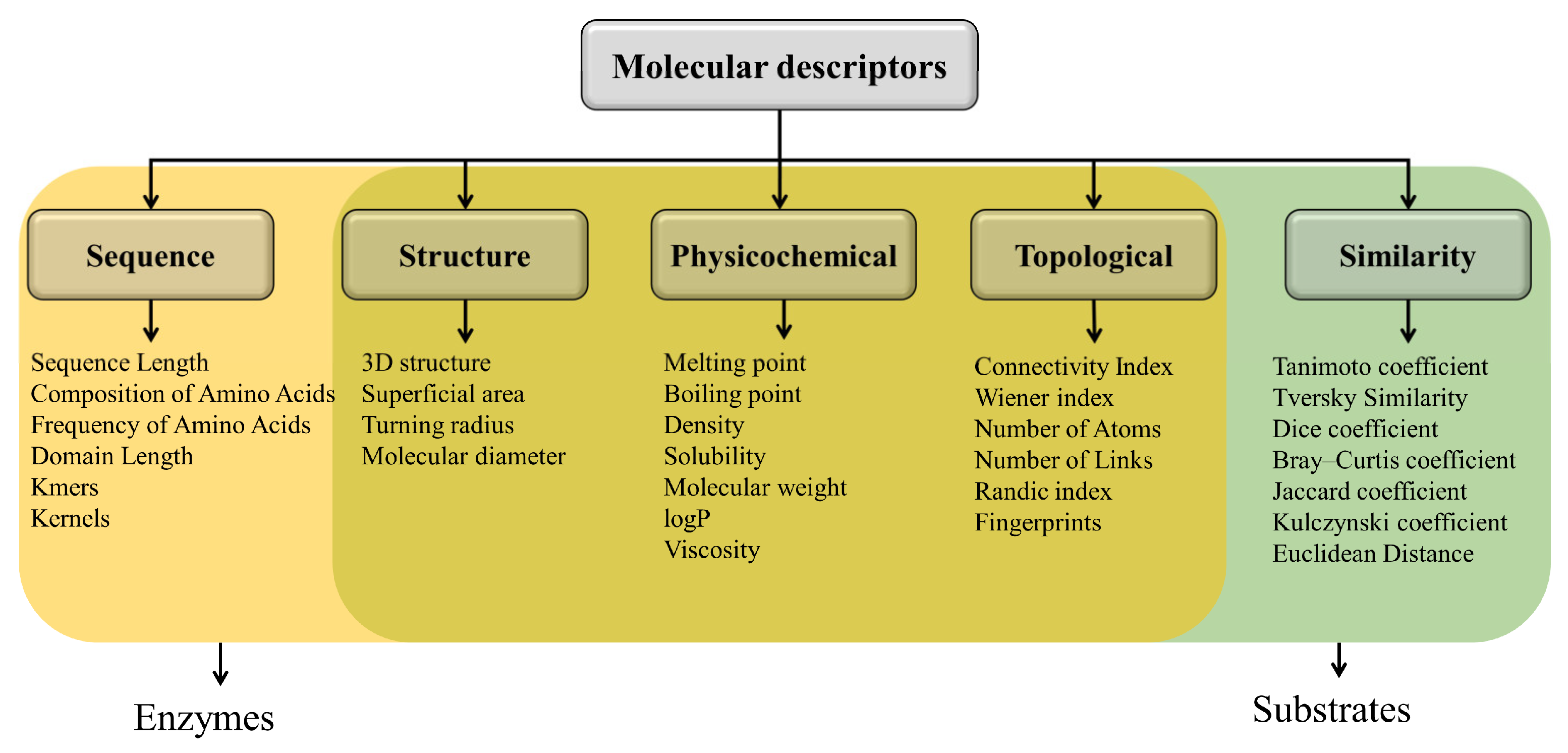
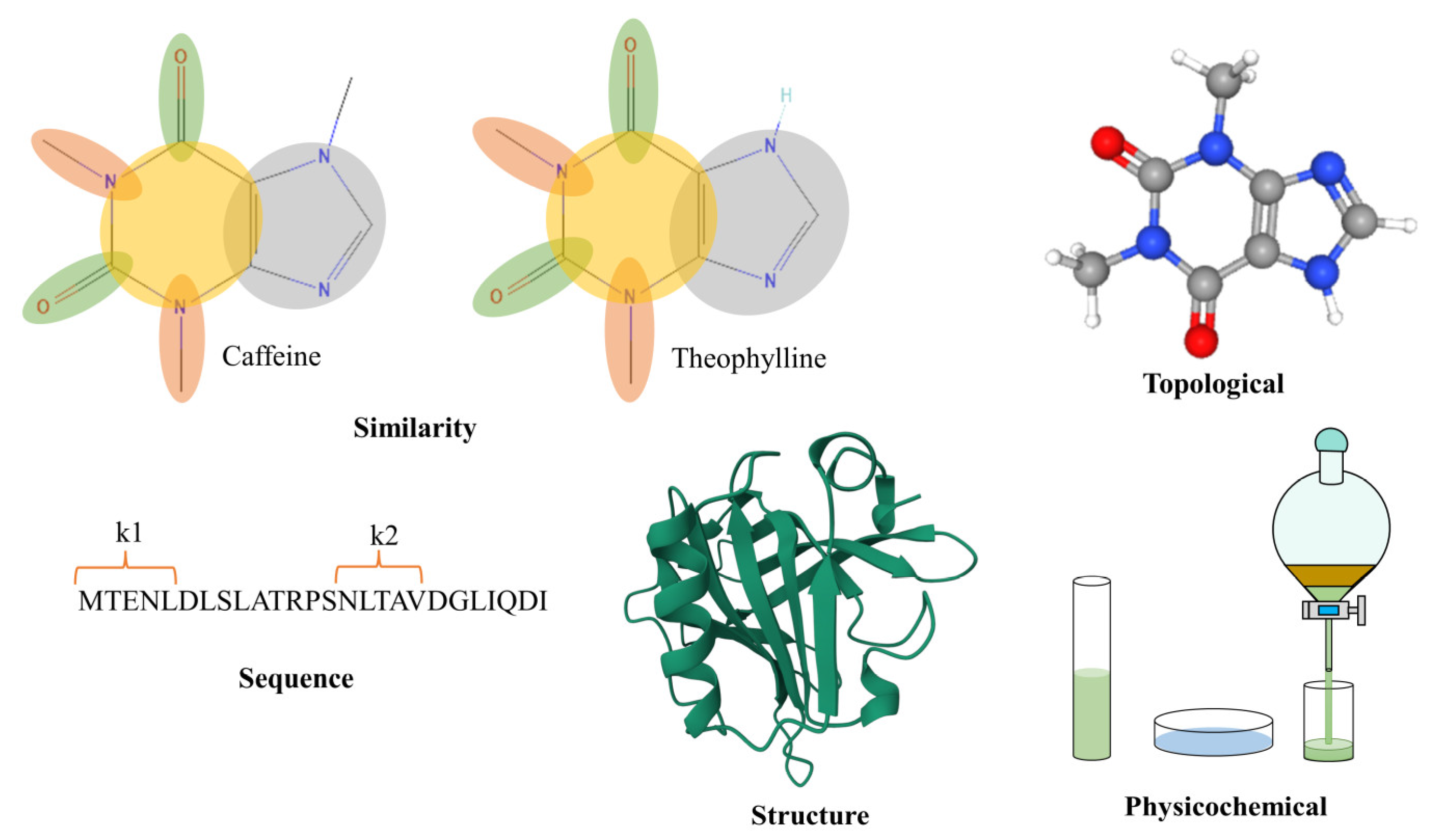
4. Characterization of Enzyme–Substrate Interactions
5. Artificial Intelligence Models
5.1. Support Vector Machine (SVM) Models
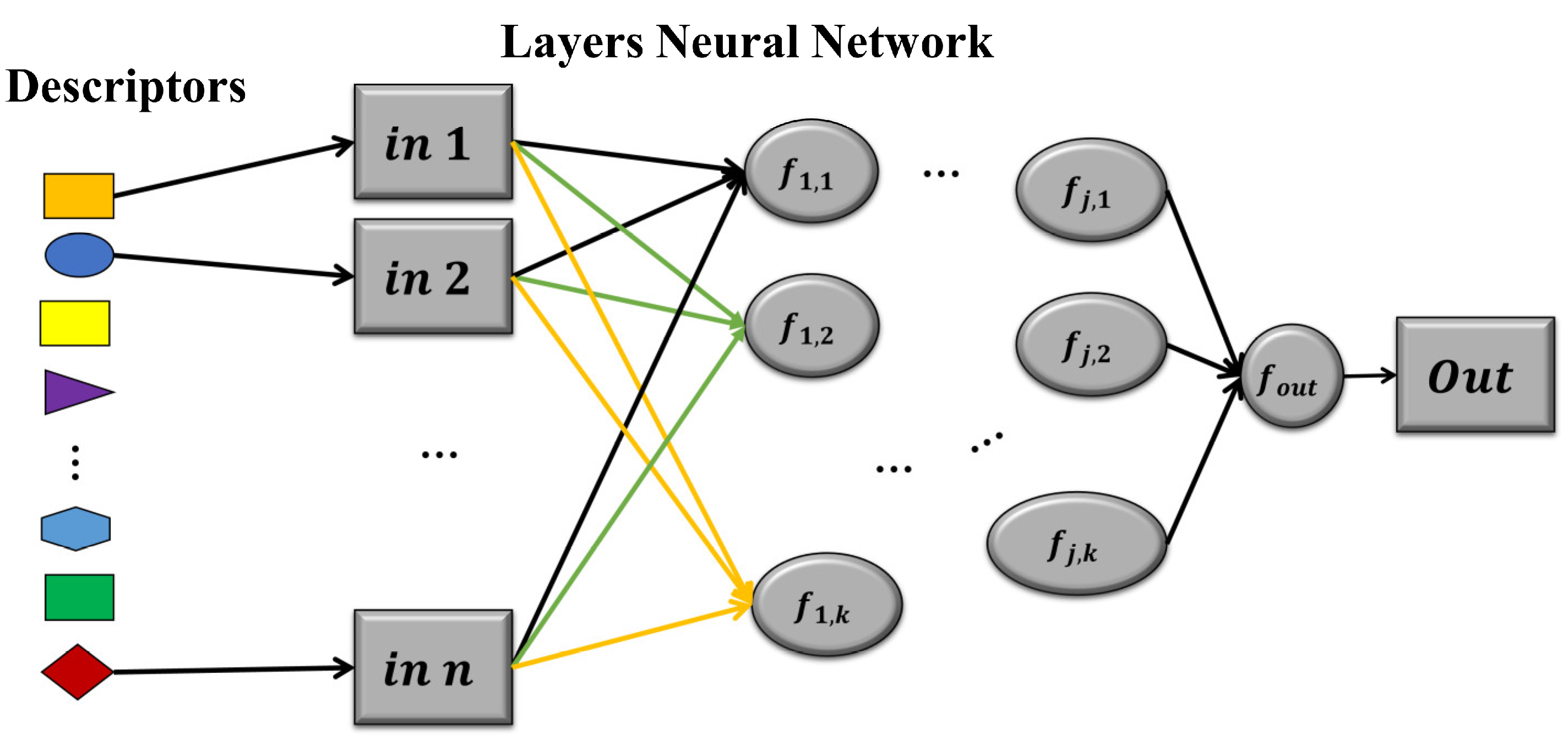
5.2. Neural Network Models
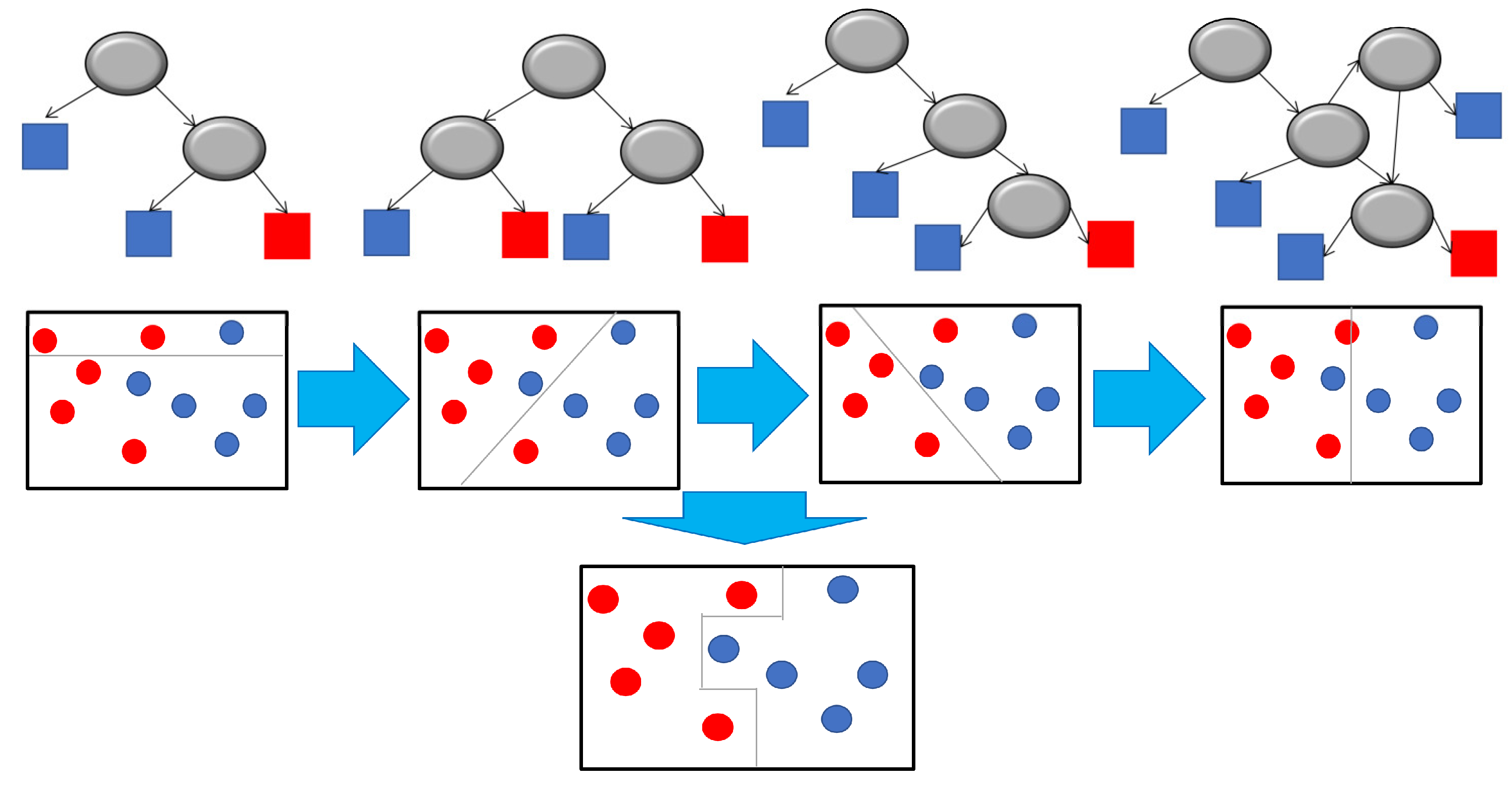
5.3. Decision Tree Models
5.4. Other Models
6. Model Validation
6.1. Cross-Validation
6.2. Out-of-Sample Validation
6.3. Experimental Validation
7. Limitations, Challenges, and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stephanopoulos, G. Synthetic Biology and Metabolic Engineering. ACS Synth. Biol. 2012, 1, 514–525. [Google Scholar] [CrossRef]
- García-Granados, R.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. Metabolic Engineering and Synthetic Biology: Synergies, Future, and Challenges. Front. Bioeng. Biotechnol. 2019, 7, 36. [Google Scholar] [CrossRef]
- Choi, K.R.; Jang, W.D.; Yang, D.; Cho, J.S.; Park, D.; Lee, S.Y. Systems Metabolic Engineering Strategies: Integrating Systems and Synthetic Biology with Metabolic Engineering. Trends Biotechnol. 2019, 37, 817–837. [Google Scholar] [CrossRef]
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223. [Google Scholar] [CrossRef]
- Banerjee, D.; Jindra, M.A.; Linot, A.J.; Pfleger, B.F.; Maranas, C.D. EnZymClass: Substrate Specificity Prediction Tool of Plant Acyl-ACP Thioesterases Based on Ensemble Learning. Curr. Res. Biotechnol. 2022, 4, 1–9. [Google Scholar] [CrossRef]
- Feehan, R.; Montezano, D.; Slusky, J.S.G. Machine Learning for Enzyme Engineering, Selection and Design. Protein Eng. Des. Sel. 2021, 34, gzab019. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.L.; Smith, M.D.; Richman, J.E.; Aukema, K.G.; Wackett, L.P. Machine Learning-Based Prediction of Activity and Substrate Specificity for OleA Enzymes in the Thiolase Superfamily. Synth. Biol. 2020, 5, ysaa004. [Google Scholar] [CrossRef]
- Du, K.-L.; Swamy, M.N.S. Fundamentals of Machine Learning. In Neural Networks and Statistical Learning; Du, K.-L., Swamy, M.N.S., Eds.; Springer: London, UK, 2014; pp. 15–65. ISBN 978-1-4471-5571-3. [Google Scholar]
- Trappenberg, T. Fundamentals of Machine Learning; Oxford University Press: Oxford, UK, 2019; ISBN 9780198828044. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Janiesch, C.; Zschech, P.; Heinrich, K. Machine Learning and Deep Learning. Electron. Mark. 2021, 31, 685–695. [Google Scholar] [CrossRef]
- Pereira, F.C.; Borysov, S.S. Chapter 2—Machine Learning Fundamentals. In Mobility Patterns, Big Data and Transport Analytics; Antoniou, C., Dimitriou, L., Pereira, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 9–29. ISBN 978-0-12-812970-8. [Google Scholar]
- Chowdhary, P. Fundamentals of Artificial Intelligence; Springer: New Delhi, India, 2020; ISBN 978-81-322-3970-3. [Google Scholar]
- Russell, S.; Norvig, P. Artificial Intelligence: A Modern Approach, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2010. [Google Scholar]
- Darwiche, A. Modeling and Reasoning with Bayesian Networks; Cambridge University Press: Cambridge, UK, 2009; ISBN 9780521884389. [Google Scholar]
- Ramos, F.T.; Cozman, F.G. Anytime Anyspace Probabilistic Inference. Int. J. Approx. Reason. 2005, 38, 53–80. [Google Scholar] [CrossRef]
- Somvanshi, M.; Chavan, P.; Tambade, S.; Shinde, S.V. A Review of Machine Learning Techniques Using Decision Tree and Support Vector Machine. In Proceedings of the 2016 International Conference on Computing Communication Control and Automation (ICCUBEA), Pune, India, 12–13 August 2016; pp. 1–7. [Google Scholar]
- Mou, Z.; Eakes, J.; Cooper, C.J.; Foster, C.M.; Standaert, R.F.; Podar, M.; Doktycz, M.J.; Parks, J.M. Machine Learning-Based Prediction of Enzyme Substrate Scope: Application to Bacterial Nitrilases. Proteins Struct. Funct. Bioinform. 2021, 89, 336–347. [Google Scholar] [CrossRef]
- Amin, S.R.; Erdin, S.; Ward, R.M.; Lua, R.C.; Lichtarge, O. Prediction and Experimental Validation of Enzyme Substrate Specificity in Protein Structures. Proc. Natl. Acad. Sci. USA 2013, 110, E4195–E4202. [Google Scholar] [CrossRef] [PubMed]
- Kroll, A.; Ranjan, S.; Engqvist, M.K.M.; Lercher, M.J. A General Model to Predict Small Molecule Substrates of Enzymes Based on Machine and Deep Learning. Nat. Commun. 2023, 14, 2787. [Google Scholar] [CrossRef] [PubMed]
- Hammoudeh, Z.; Lowd, D. Training Data Influence Analysis and Estimation: A Survey. arXiv 2023, arXiv:2212.04612. [Google Scholar]
- Paullada, A.; Raji, I.D.; Bender, E.M.; Denton, E.; Hanna, A. Data and Its (Dis)Contents: A Survey of Dataset Development and Use in Machine Learning Research. Patterns 2021, 2, 100336. [Google Scholar] [CrossRef]
- Gudivada, V.N.; Irfan, M.T.; Fathi, E.; Rao, D.L. Chapter 5—Cognitive Analytics: Going Beyond Big Data Analytics and Machine Learning. In Handbook of Statistics; Gudivada, V.N., Raghavan, V.V., Govindaraju, V., Rao, C.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 35, pp. 169–205. ISBN 0169-7161. [Google Scholar]
- Chen, H.; Li, T.; Fan, X.; Luo, C. Feature Selection for Imbalanced Data Based on Neighborhood Rough Sets. Inf. Sci. 2019, 483, 1–20. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J. The Impact of Imbalanced Training Data on Machine Learning for Author Name Disambiguation. Scientometrics 2018, 117, 511–526. [Google Scholar] [CrossRef]
- Narwane, S.; Sawarkar, S. Machine Learning and Class Imbalance: A Literature Survey. Ind. Eng. J. 2019, 12. [Google Scholar] [CrossRef]
- Dean, J.; Corrado, G.; Monga, R.; Chen, K.; Devin, M.; Mao, M.; Ranzato, M.; Senior, A.; Tucker, P.; Yang, K.; et al. Large Scale Distributed Deep Networks. In Advances in Neural Information Processing Systems; Pereira, F., Burges, C.J., Bottou, L., Weinberger, K.Q., Eds.; Curran Associates, Inc.: Red Hook, NY, USA, 2012; Volume 25. [Google Scholar]
- Li, F.; Yuan, L.; Lu, H.; Li, G.; Chen, Y.; Engqvist, M.K.M.; Kerkhoven, E.J.; Nielsen, J. Deep Learning-Based Kcat Prediction Enables Improved Enzyme-Constrained Model Reconstruction. Nat. Catal. 2022, 5, 662–672. [Google Scholar] [CrossRef]
- Koutsandreas, T.; Pilalis, E.; Chatziioannou, A. A Machine-Learning Approach for Theof Enzymatic Activity of Proteins in Metagenomic Samples. In Artificial Intelligence Applications and Innovations, Proceedings of the IFIP International Conference on Artificial Intelligence Applications and Innovations, Paphos, Cyprus, 30 September–2 October 2013; Papadopoulos, H., Andreou, A.S., Iliadis, L., Maglogiannis, I., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 81–87. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H. Protein Sequence Databases. Curr. Opin. Chem. Biol. 2004, 8, 76–80. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- wwPDB consortium. Protein Data Bank: The Single Global Archive for 3D Macromolecular Structure Data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc Database of Metabolic Pathways and Enzymes—A 2019 Update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR Core Data Resource in 2021: New Developments and Updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG Mapping Tools for Uncovering Hidden Features in Biological Data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI Reference Sequences (RefSeq): Current Status, New Features and Genome Annotation Policy. Nucleic Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef]
- Karp, P.D.; Riley, M.; Saier, M.; Paulsen, I.T.; Paley, S.M.; Pellegrini-Toole, A. The EcoCyc and MetaCyc Databases. Nucleic Acids Res. 2000, 28, 56–59. [Google Scholar] [CrossRef]
- Taheri, K.; Moradi, H.; Tavassolipour, M. Collaboration Graph for Feature Set Partitioning in Data Classification. Expert Syst. Appl. 2023, 213, 118988. [Google Scholar] [CrossRef]
- Cordeiro de Amorim, R. Unsupervised Feature Selection for Large Data Sets. Pattern Recognit. Lett. 2019, 128, 183–189. [Google Scholar] [CrossRef]
- Wang, J.; Yang, B.; Revote, J.; Leier, A.; Marquez-Lago, T.T.; Webb, G.; Song, J.; Chou, K.-C.; Lithgow, T. POSSUM: A Bioinformatics Toolkit for Generating Numerical Sequence Feature Descriptors Based on PSSM Profiles. Bioinformatics 2017, 33, 2756–2758. [Google Scholar] [CrossRef]
- Dong, L.; Wang, R.; Chen, D. Incremental Feature Selection with Fuzzy Rough Sets for Dynamic Data Sets. Fuzzy Sets Syst. 2023, 467, 108503. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Dhawan, A. Chapter 5—In Silico Approaches for Predictive Toxicology. In In Vitro Toxicology; Dhawan, A., Kwon, S., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 91–109. ISBN 978-0-12-804667-8. [Google Scholar]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Chapter 21—Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. In Dosage Form Design Parameters; Tekade, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 731–755. ISBN 978-0-12-814421-3. [Google Scholar]
- Ben-Hur, A.; Ong, C.S.; Sonnenburg, S.; Schölkopf, B.; Rätsch, G. Support Vector Machines and Kernels for Computational Biology. PLoS Comput. Biol. 2008, 4, e1000173. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Chou, L.-C.; Chang, D.T.-H. Predicting Protein-Protein Interactions in Unbalanced Data Using the Primary Structure of Proteins. BMC Bioinform. 2010, 11, 167. [Google Scholar] [CrossRef]
- Saigo, H.; Vert, J.-P.; Ueda, N.; Akutsu, T. Protein Homology Detection Using String Alignment Kernels. Bioinformatics 2004, 20, 1682–1689. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef]
- Çamoǧlu, O.; Can, T.; Singh, A.K.; Wang, Y.-F. Decision Tree Based Information Integration for Automated Protein Classification. J. Bioinform. Comput. Biol. 2005, 3, 717–742. [Google Scholar] [CrossRef]
- Kroll, A.; Engqvist, M.; Heckmann, D.; Lercher, M. Deep Learning Allows Genome-Scale Prediction of Michaelis Constants from Structural Features. PLoS Biol. 2021, 19, e3001402. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph Neural Networks: A Review of Methods and Applications. AI Open 2020, 1, 57–81. [Google Scholar] [CrossRef]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef]
- Prince, S.J.D. Understanding Deep Learning; The MIT Press: Cambridge, MA, USA, 2023; ISBN 9780262048644. [Google Scholar]
- Alaskar, H.; Saba, T. Machine Learning and Deep Learning: A Comparative Review. In Proceedings of Integrated Intelligence Enable Networks and Computing; Springer: Singapore, 2021; pp. 143–150. ISBN 978-981-33-6306-9. [Google Scholar]
- Deshpande, M.; Karypis, G. Evaluation of Techniques for Classifying Biological Sequences. In Proceedings of the Advances in Knowledge Discovery and Data Mining, Tainan, Taiwan, 13–16 May 2014; Chen, M.-S., Yu, P.S., Liu, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 417–431. [Google Scholar]
- Xing, Z.; Pei, J.; Keogh, E. A Brief Survey on Sequence Classification. SIGKDD Explor. Newsl. 2010, 12, 40–48. [Google Scholar] [CrossRef]
- Goldman, S.; Das, R.; Yang, K.K.; Coley, C.W. Machine Learning Modeling of Family Wide Enzyme-Substrate Specificity Screens. PLoS Comput. Biol. 2022, 18, e1009853. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Upadhyay, V.; Boorla, V.S.; Maranas, C.D. Rank-Ordering of Known Enzymes as Starting Points for Re-Engineering Novel Substrate Activity Using a Convolutional Neural Network. Metab. Eng. 2023, 78, 171–182. [Google Scholar] [CrossRef]
- Costa, E.P.; Lorena, A.C.; Carvalho, A.C.P.L.F.; Freitas, A.A.; Holden, N. Comparing Several Approaches for Hierarchical Classification of Proteins with Decision Trees. In Advances in Bioinformatics and Computational Biology, Proceedings of the Second Brazilian Symposium on Bioinformatics, BSB 2007, Angra dos Reis, Brazil, 29–31 August 2007; Sagot, M.-F., Walter, M.E.M.T., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 126–137. [Google Scholar]
- Ebrahimi, M. Sequence-Based Prediction of Enzyme Thermostability Through Bioinformatics Algorithms. Curr. Bioinform. 2010, 5, 195–203. [Google Scholar] [CrossRef]
- Si, S.; Zhang, H.; Keerthi, S.S.; Mahajan, D.; Dhillon, I.S.; Hsieh, C.-J. Gradient Boosted Decision Trees for High Dimensional Sparse Output. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; Volume 70, pp. 3182–3190. [Google Scholar]
- Feehan, R.; Franklin, M.W.; Slusky, J.S.G. Machine Learning Differentiates Enzymatic and Non-Enzymatic Metals in Proteins. Nat. Commun. 2021, 12, 3712. [Google Scholar] [CrossRef]
- Leslie, C.; Eskin, E.; Noble, W.S. The spectrum kernel: A string kernel for svm protein classification. In Biocomputing; World Scientific Publishing: Singapore, 2002; pp. 564–575. [Google Scholar]
- Muda, H.M.; Saad, P.; Othman, R.M. Remote Protein Homology Detection and Fold Recognition Using Two-Layer Support Vector Machine Classifiers. Comput. Biol. Med. 2011, 41, 687–699. [Google Scholar] [CrossRef]
- Alzubaidi, L.; Zhang, J.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaría, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of Deep Learning: Concepts, CNN Architectures, Challenges, Applications, Future Directions. J. Big Data 2021, 8, 53. [Google Scholar] [CrossRef]
- Mittal, R.; Arora, S.; Bansal, V.; Bhatia, M.P.S. An Extensive Study on Deep Learning: Techniques, Applications. Arch. Comput. Methods Eng. 2021, 28, 4471–4485. [Google Scholar] [CrossRef]
- Li, S.; Wan, F.; Shu, H.; Jiang, T.; Zhao, D.; Zeng, J. MONN: A Multi-Objective Neural Network for Predicting Compound-Protein Interactions and Affinities. Cell Syst. 2020, 10, 308–322.e11. [Google Scholar] [CrossRef]
- Wu, K.E.; Yang, K.K.; van den Berg, R.; Alamdari, S.; Zou, J.Y.; Lu, A.X.; Amini, A.P. Protein Structure Generation via Folding Diffusion. Nat. Commun. 2024, 15, 1059. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, J.; Wang, Y.; Chen, M.; Wang, D.; Xu, D.; Cheng, J. Diffusion Models in Bioinformatics: A New Wave of Deep Learning Revolution in Action. arXiv 2023, arXiv:2302.10907. [Google Scholar]
- Corso, G.; Stärk, H.; Bowen, J.; Barzilay, R.; Jaakkola, T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. arXiv 2023, arXiv:2210.01776. [Google Scholar]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De Novo Design of Protein Structure and Function with RFdiffusion. Nature 2023, 620, 1089–1100. [Google Scholar] [CrossRef]
- Canela, M.Á.; Alegre, I.; Ibarra, A. Out-of-Sample Validation. In Quantitative Methods for Management: A Practical Approach; Springer International Publishing: Cham, Switzerland, 2019; pp. 83–89. ISBN 978-3-030-17554-2. [Google Scholar]
- Repecka, D.; Jauniskis, V.; Karpus, L.; Rembeza, E.; Rokaitis, I.; Zrimec, J.; Poviloniene, S.; Laurynenas, A.; Viknander, S.; Abuajwa, W.; et al. Expanding Functional Protein Sequence Spaces Using Generative Adversarial Networks. Nat. Mach. Intell. 2021, 3, 324–333. [Google Scholar] [CrossRef]
- Sokolova, M.; Japkowicz, N.; Szpakowicz, S. Beyond Accuracy, F-Score and ROC: A Family of Discriminant Measures for Performance Evaluation. In Advances in Artificial Intelligence, Proceedings of the AI 2006: Advances in Artificial Intelligence, Hobart, Australia, 4–8 December 2006; Sattar, A., Kang, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1015–1021. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Type of Information | Quantity of Proteins | Main Focus | Strengths | Weaknesses | Ref. |
|---|---|---|---|---|---|---|
| UniProt | Sequences, functions, and structures | 248,272,897 structures (569,793 reviewed) | Proteins and their attributes | Wide coverage and comprehensive and updated information | Redundant and unreviewed data | [30,31] |
| PDB | Structural information | 208,066 PDB structures + 1,068,577 computerized structure models | 3D structures of proteins and enzymes | Revised and non-redundant database | Focus only on structure | [32] |
| BRENDA | Functional and metabolic information | 32,832,265 sequences, 90,000 enzymes, and 13,000 organisms | Enzymes, their reactions, and biochemical properties | Database specialized in enzymes, their function, biochemical properties, and reactions; revised database | Slow updates; requires prior knowledge in biochemistry and molecular biology | [34] |
| KEGG | Information on metabolic pathways and gene/protein functions | 1,098,631 metabolic pathways and 49,962,693 genes | Metabolic pathways and gene functions | Interconnection with other databases | Requires prior knowledge in biochemistry and molecular biology | [35] |
| NCBI | Protein sequences, structures, gene sequences, and annotations | 40,000,000 | Various protein information | Interconnection with other databases | Redundant information | [36] |
| MetaCyc | Metabolic pathways and enzymes | >2749 pathways | Metabolic pathways and enzymes from different organisms | Revised and non-redundant database | It is limited to metabolic pathways and enzymes | [33,37] |
| Main Approach | Type of Descriptors | Data Used | References |
|---|---|---|---|
| Prediction of range of substrates in bacterial nitrilases | Sequence, physicochemical, and structure | Experimental activity data, alignments, electrostatic potential, and 3D substrate structure | [18] |
| Detection of functional similarities | Sequence and structure | Alignments, sequences, and structures | [19] |
| Discrimination of substrate function | Sequence and structure | Alignments and fingerprints | [45,46] |
| Approach based on fingerprints and properties: Michaelis constant prediction | Similarity and topology | Fingerprints, molecular weight, LogP, and others | [49] |
| Comparison of results and new approaches | Similarity and topology | Fingerprints and MPNN | [29] |
| Algorithm | Application | Performance | References |
|---|---|---|---|
| Support vector machines (SVMs) | Enzymatic and substrate classification and prediction | 80% accuracy | [5] |
| Support vector machines (SVMs) and kernel techniques | Analysis and processing of complex biological data | 77–91.4% accuracy | [44] |
| Decision trees | Differentiation of metals in proteins | 94.2% accuracy | [6] |
| Neural networks | Classification of enzymes, substrates, and sequences | >85% accuracy | [4,54,55] |
| Neural networks | Prediction of enzyme–substrate interactions | >73.2% | [5,7,20] |
| Neural networks | Prediction of enzyme specificity | Complex relationship capture | [56] |
| Neural networks | Prediction of protein structures and interactions | Capture of complex features | [57] |
| Convolutional neural networks | Enzyme classification and redesign | 80.72% accuracy | [58] |
| Decision trees | Protein classification and regression | 62% | [59,60] |
| Gradient augmentation trees | Prediction of enzyme activity | 79% | [5,61] |
| Random forest, feedforward neural network, and Naive Bayes | Prediction of OleA enzyme activity and specificity | 82.6%, 73.2%, 58.6% | [7] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salas-Nuñez, L.F.; Barrera-Ocampo, A.; Caicedo, P.A.; Cortes, N.; Osorio, E.H.; Villegas-Torres, M.F.; González Barrios, A.F. Machine Learning to Predict Enzyme–Substrate Interactions in Elucidation of Synthesis Pathways: A Review. Metabolites 2024, 14, 154. https://doi.org/10.3390/metabo14030154
Salas-Nuñez LF, Barrera-Ocampo A, Caicedo PA, Cortes N, Osorio EH, Villegas-Torres MF, González Barrios AF. Machine Learning to Predict Enzyme–Substrate Interactions in Elucidation of Synthesis Pathways: A Review. Metabolites. 2024; 14(3):154. https://doi.org/10.3390/metabo14030154
Chicago/Turabian StyleSalas-Nuñez, Luis F., Alvaro Barrera-Ocampo, Paola A. Caicedo, Natalie Cortes, Edison H. Osorio, Maria F. Villegas-Torres, and Andres F. González Barrios. 2024. "Machine Learning to Predict Enzyme–Substrate Interactions in Elucidation of Synthesis Pathways: A Review" Metabolites 14, no. 3: 154. https://doi.org/10.3390/metabo14030154
APA StyleSalas-Nuñez, L. F., Barrera-Ocampo, A., Caicedo, P. A., Cortes, N., Osorio, E. H., Villegas-Torres, M. F., & González Barrios, A. F. (2024). Machine Learning to Predict Enzyme–Substrate Interactions in Elucidation of Synthesis Pathways: A Review. Metabolites, 14(3), 154. https://doi.org/10.3390/metabo14030154