
Integration of Metabolome and Transcriptome Reveals the Major Metabolic Pathways and Potential Biomarkers in Response to Freeze-Stress Regulation in Apple (Malus domestica)


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Measurement of Physiological Indexes and Recovery Culture of Plant Materials
2.3. Metabolomic and Bioinformatic Analysis
2.4. Transcriptomics and Bioinformatics Analysis
2.5. Detection of Relative Gene Expression
3. Results
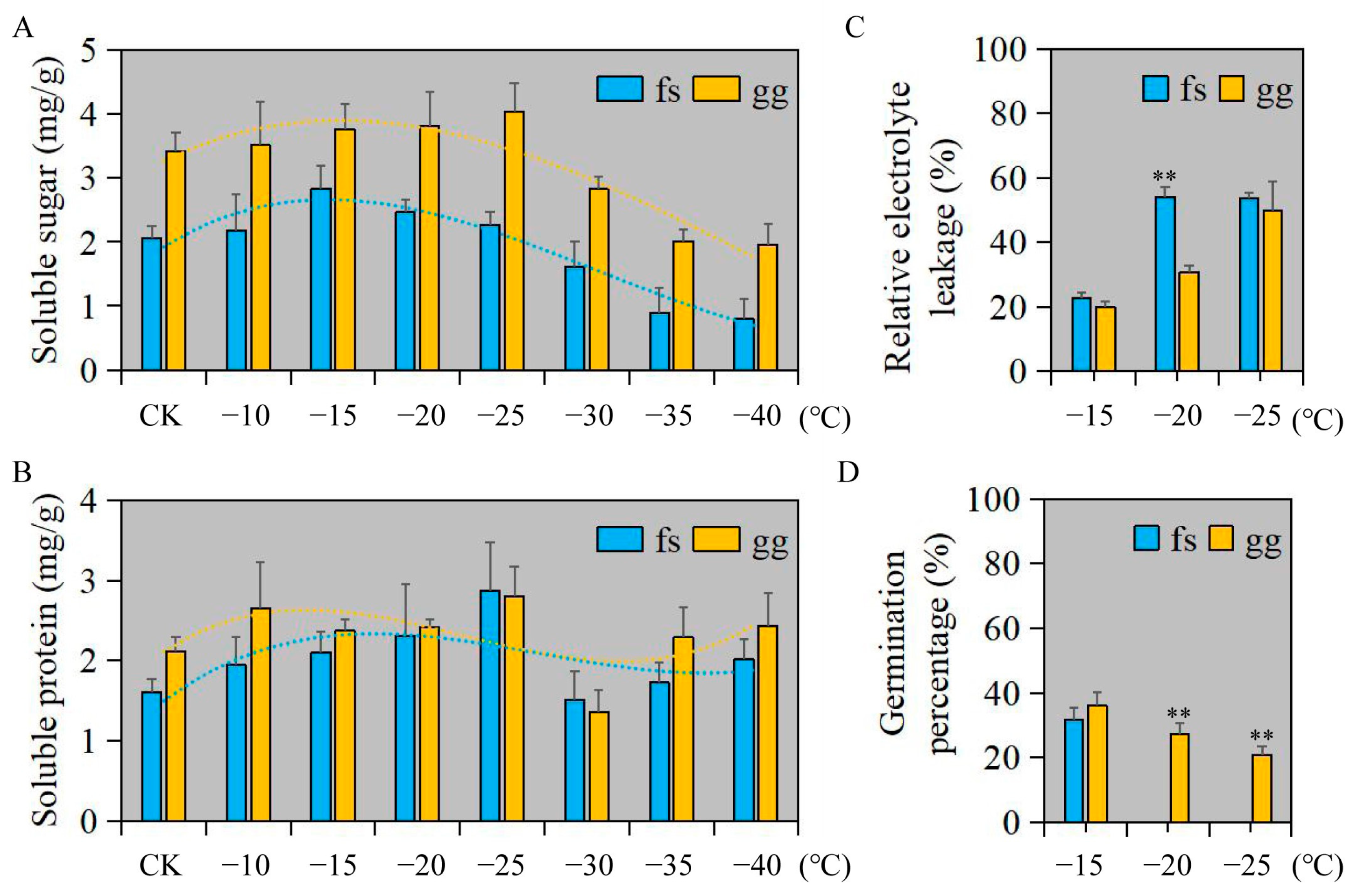
3.1. Physiological Index Evaluation of Apple under Freezing Stress
3.2. Metabolome Profiling of Forty-Eight Apple Samples
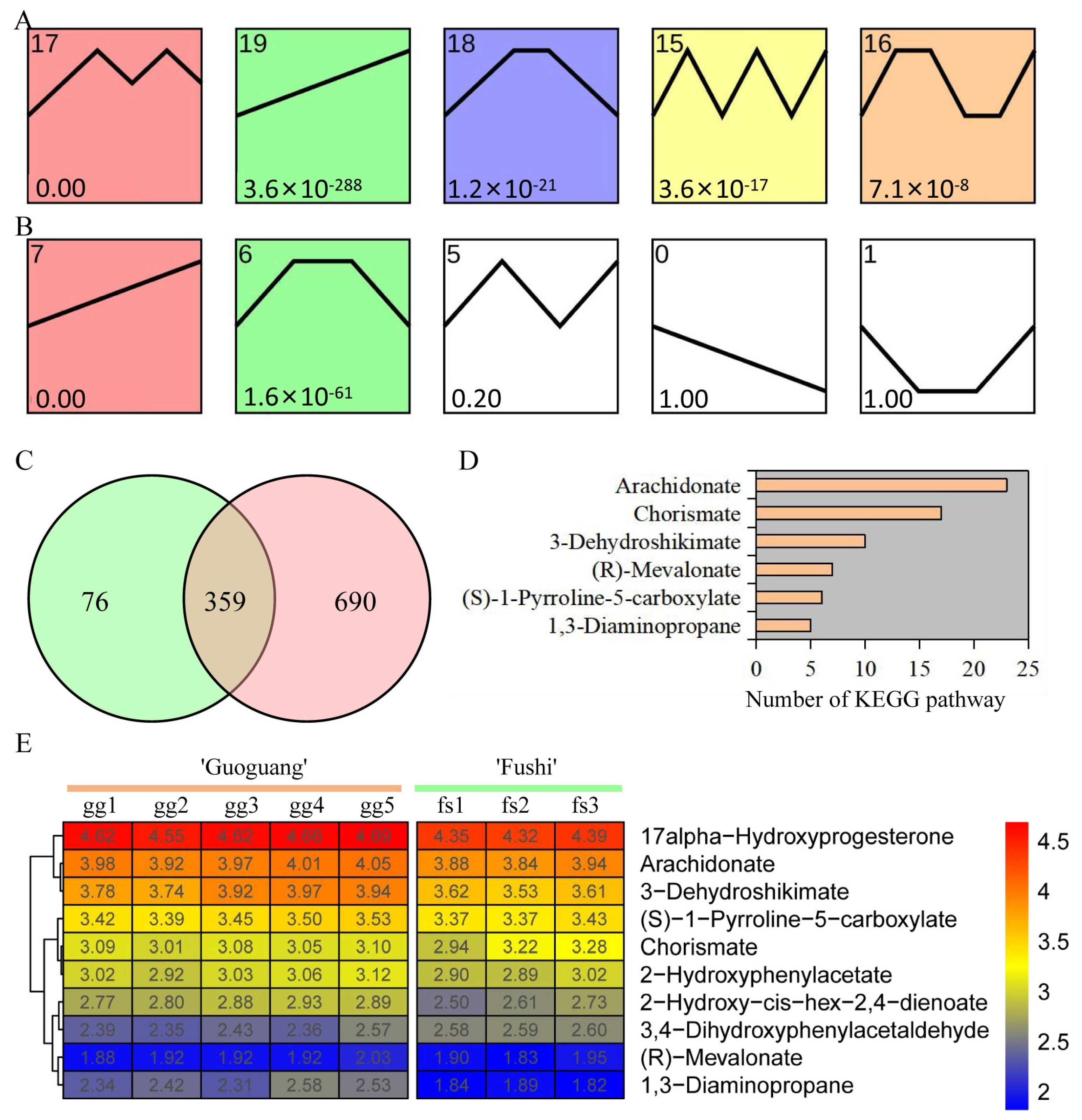
3.3. Identification and Functional Enrichment of Differentially Accumulated Metabolites
3.4. Trend of Metabolite Content under Low Temperature Treatment
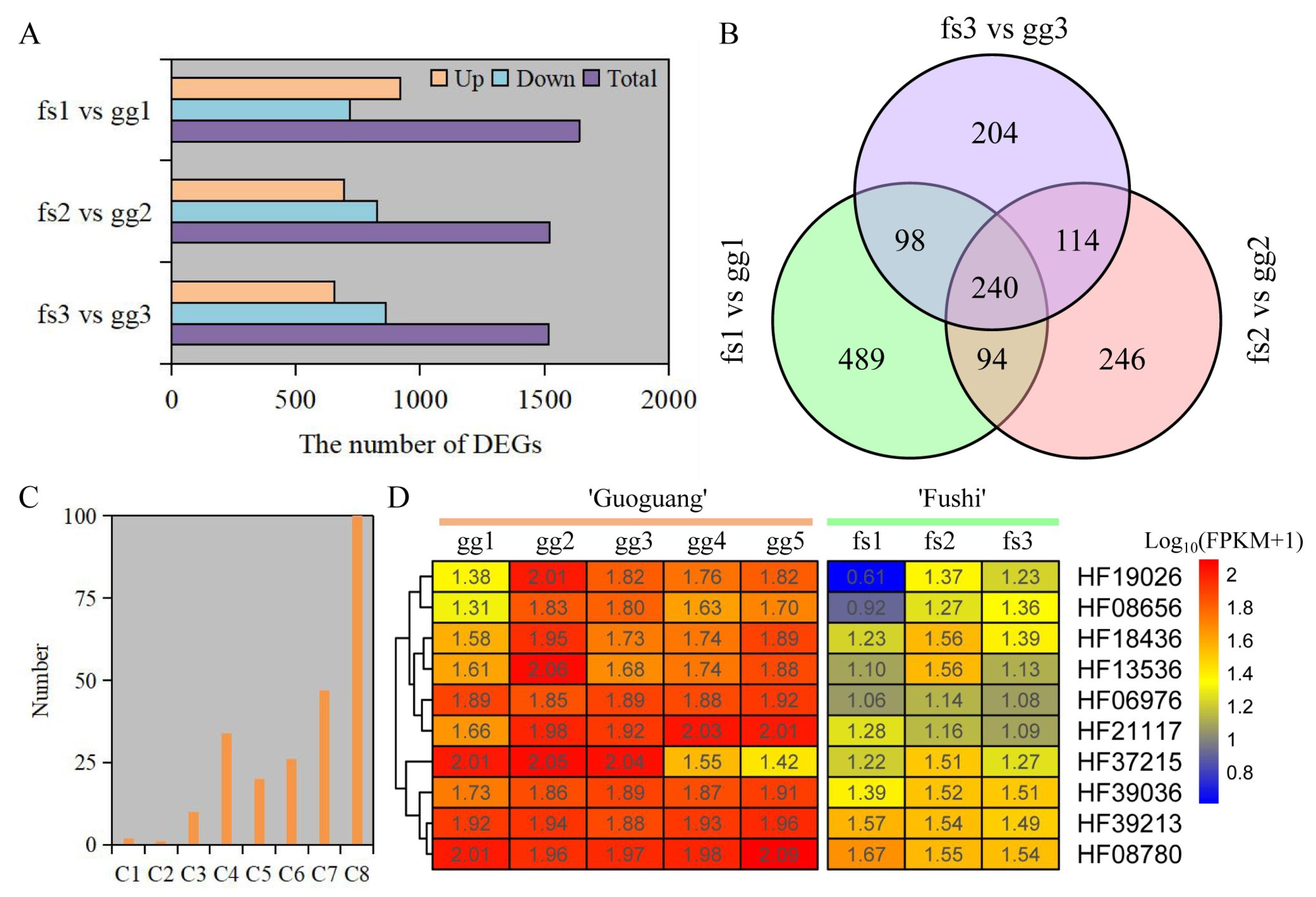
3.5. Transcriptome Profiling and DEGs Analysis
3.6. GO and KEGG Analysis of DEGs
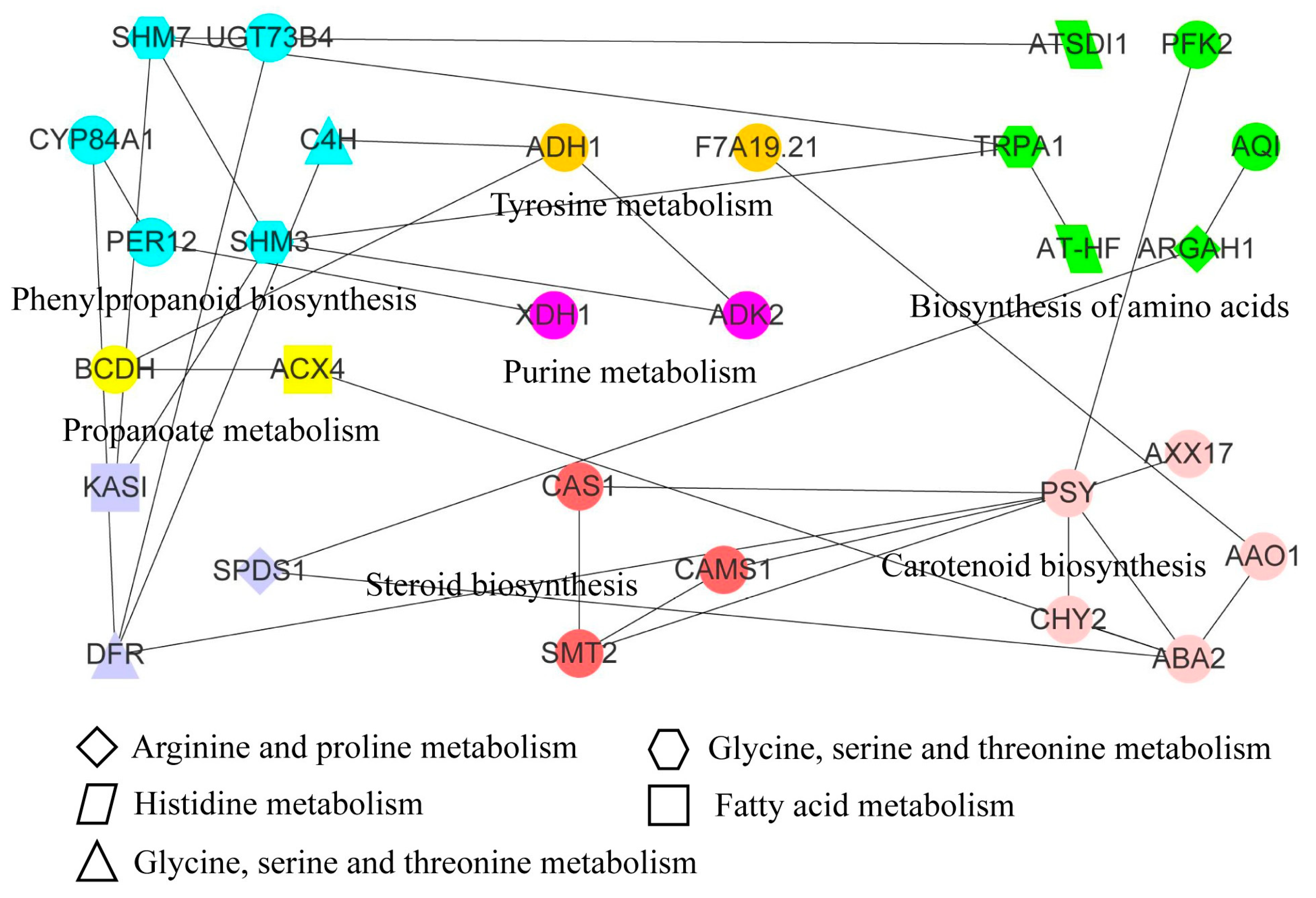
3.7. Transcriptomic and Metabolomic Analysis of Apple under Freezing Stress
3.8. Analysis of Phenylpropanoid Biosynthesis Pathway in Apples under Low Temperature Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, D.; Uemura, M.; Kawamura, Y. Freezing Tolerance of Plant Cells: From the Aspect of Plasma Membrane and Microdomain. In Survival Strategies in Extreme Cold and Desiccation: Adaptation Mechanisms and Their Applications; Iwaya-Inoue, M., Sakurai, M., Uemura, M., Eds.; Springer: Singapore, 2018; pp. 61–79. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Chong, K.; Xu, Y. Chilling tolerance in rice: Past and present. J. Plant Physiol. 2022, 268, 153576. [Google Scholar] [CrossRef]
- Villouta, C.; Workmaster, B.A.; Bolivar-Medina, J.; Sinclair, S.; Atucha, A. Freezing stress survival mechanisms in Vaccinium macrocarpon Ait. terminal buds. Tree Physiol. 2020, 40, 841–855. [Google Scholar] [CrossRef]
- Charrier, G.; Ngao, J.; Saudreau, M.; Ameglio, T. Effects of environmental factors and management practices on microclimate, winter physiology, and frost resistance in trees. Front. Plant Sci. 2015, 6, 259. [Google Scholar] [CrossRef]
- Neuner, G. Frost resistance in alpine woody plants. Front. Plant Sci. 2014, 5, 654. [Google Scholar] [CrossRef]
- Webb, M.S.; Uemura, M.; Steponkus, P.L. A Comparison of Freezing Injury in Oat and Rye: Two Cereals at the Extremes of Freezing Tolerance. Plant Physiol. 1994, 104, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Maurya, J.P.; Bhalerao, R.P. Photoperiod- and temperature-mediated control of growth cessation and dormancy in trees: A molecular perspective. Ann. Bot. 2017, 120, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Bratzel, F.; Turck, F. Molecular memories in the regulation of seasonal flowering: From competence to cessation. Genome Biol. 2015, 16, 192. [Google Scholar] [CrossRef] [PubMed]
- Ambroise, V.; Legay, S.; Guerriero, G.; Hausman, J.-F.; Cuypers, A.; Sergeant, K. The Roots of Plant Frost Hardiness and Tolerance. Plant Cell Physiol. 2020, 61, 3–20. [Google Scholar] [CrossRef]
- Hincha, D.K.; Zuther, E. Plant Cold Acclimation and Freezing Tolerance. In Plant Cold Acclimation: Methods and Protocols; Hincha, D.K., Zuther, E., Eds.; Springer: New York, NY, USA, 2014; pp. 1–6. [Google Scholar] [CrossRef]
- Morales, M.; Munné-Bosch, S. Oxidative Stress: A Master Regulator of Plant Trade-Offs? Trends Plant Sci. 2016, 21, 996–999. [Google Scholar] [CrossRef]
- Tylewicz, S.; Petterle, A.; Marttila, S.; Miskolczi, P.; Azeez, A.; Singh, R.K.; Immanen, J.; Mähler, N.; Hvidsten, T.R.; Eklund, D.M.; et al. Photoperiodic control of seasonal growth is mediated by ABA acting on cell-cell communication. Science 2018, 360, 212–215. [Google Scholar] [CrossRef]
- Friedrich, T.; Faivre, L.; Bäurle, I.; Schubert, D. Chromatin-based mechanisms of temperature memory in plants. Plant Cell Environ. 2019, 42, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.; Guy, C.L. β-Amylase Induction and the Protective Role of Maltose during Temperature Shock. Plant Physiol. 2004, 135, 1674–1684. [Google Scholar] [CrossRef]
- Rohde, P.; Hincha, D.K.; Heyer, A.G. Heterosis in the freezing tolerance of crosses between two Arabidopsis thaliana accessions (Columbia-0 and C24) that show differences in non-acclimated and acclimated freezing tolerance. Plant J. 2004, 38, 790–799. [Google Scholar] [CrossRef]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Fowler, S.; Fiehn, O.; Thomashow, M.F. A prominent role for the CBF cold response pathway in configuring the low-temperature metabolome of Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 15243–15248. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zheng, G.; Yu, X.; Liu, S.; Dong, X.; Cao, X.; Fang, X.; Li, H.; Jin, J.; Mi, W.; et al. Comparative Transcriptomics and Proteomics Analyses of Leaves Reveals a Freezing Stress-Responsive Molecular Network in Winter Rapeseed (Brassica rapa L.). Front. Plant Sci. 2021, 12, 664311. [Google Scholar] [CrossRef]
- Zhou, P.; Li, X.; Liu, X.; Wen, X.; Zhang, Y.; Zhang, D. Transcriptome profiling of Malus sieversii under freezing stress after being cold-acclimated. BMC Genom. 2021, 22, 681. [Google Scholar] [CrossRef]
- Han, Z.; Xu, X.; Zhang, S.; Zhao, Q.; Li, H.; Cui, Y.; Li, X.; Wang, L.; Chen, S.; Zhao, X. Transcriptomics Profiling of Acer pseudosieboldianum Molecular Mechanism against Freezing Stress. Int. J. Mol. Sci. 2022, 23, 14676. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.; Kopka, J.; Sung, D.Y.; Zhao, W.; Popp, M.; Porat, R.; Guy, C.L. Transcript and metabolite profiling during cold acclimation of Arabidopsis reveals an intricate relationship of cold-regulated gene expression with modifications in metabolite content. Plant J. 2007, 50, 967–981. [Google Scholar] [CrossRef]
- Kou, S.; Chen, L.; Tu, W.; Scossa, F.; Wang, Y.; Liu, J.; Fernie, A.R.; Song, B.; Xie, C. The arginine decarboxylase gene ADC1, associated to the putrescine pathway, plays an important role in potato cold-acclimated freezing tolerance as revealed by transcriptome and metabolome analyses. Plant J. 2018, 96, 1283–1298. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, Y.; Zuo, W.T.; Gao, Y.R.; Li, R.Z.; Yu, C.X.; Liu, Z.Y.; Zheng, Y.; Shen, Y.Y.; Duan, L.S. Integration of Metabolome and Transcriptome Studies Reveals Flavonoids, Abscisic Acid, and Nitric Oxide Comodulating the Freezing Tolerance in Liriope spicata. Front. Plant Sci. 2021, 12, 764625. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Fang, J.; Lin, M.; Hu, C.; Qi, X.; Chen, J.; Zhong, Y.; Muhammad, A.; Li, Z.; Li, Y. Comparative Metabolomic and Transcriptomic Studies Reveal Key Metabolism Pathways Contributing to Freezing Tolerance under Cold Stress in Kiwifruit. Front. Plant Sci. 2021, 12, 628969. [Google Scholar] [CrossRef]
- Li, P.; Zheng, T.; Li, L.; Liu, W.; Qiu, L.; Ahmad, S.; Wang, J.; Cheng, T.; Zhang, Q. Integration of chromatin accessibility and gene expression reveals new regulators of cold hardening to enhance freezing tolerance in Prunus mume. J. Exp. Bot. 2023, 74, 2173–2187. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Takagi, T.; Nakamura, M.; Hayashi, H.; Inatsugi, R.; Yano, R.; Nishida, I. The Leaf-Order-Dependent Enhancement of Freezing Tolerance in Cold-Acclimated Arabidopsis Rosettes is not Correlated with the Transcript Levels of the Cold-Inducible Transcription Factors of CBF/DREB1. Plant Cell Physiol. 2003, 44, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Huang, Y.; Zhou, M.; Zhang, G. The influence of cold acclimation on antioxidative enzymes and antioxidants in sensitive and tolerant barley cultivars. Biol. Plant. 2009, 53, 257–262. [Google Scholar] [CrossRef]
- Guo, X.; Liu, D.; Chong, K. Cold signaling in plants: Insights into mechanisms and regulation. J. Integr. Plant Biol. 2018, 60, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ding, Y.; Yang, S. Molecular Regulation of CBF Signaling in Cold Acclimation. Trends Plant Sci. 2018, 23, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, D.; Komatsu, K.; Sakata, Y. ABA in bryophytes: How a universal growth regulator in life became a plant hormone? J. Plant Res. 2011, 124, 437–453. [Google Scholar] [CrossRef]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, Y.; Shi, Y.; Zhang, X.; Zhang, S.; Gong, Z.; Yang, S. MPK3- and MPK6-Mediated ICE1 Phosphorylation Negatively Regulates ICE1 Stability and Freezing Tolerance in Arabidopsis. Dev. Cell 2017, 43, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, T.; Shen, X.; Liu, J.; Zhao, D.; Sun, Y.; Wang, L.; Liu, Y.; Gong, X.; Liu, Y.; et al. Serum metabolomics for early diagnosis of esophageal squamous cell carcinoma by UHPLC-QTOF/MS. Metabolomics 2016, 12, 116. [Google Scholar] [CrossRef]
- Shoko, T.; Maharaj, V.J.; Naidoo, D.; Tselanyane, M.; Nthambeleni, R.; Khorombi, E.; Apostolides, Z. Anti-aging potential of extracts from Sclerocarya birrea (A. Rich.) Hochst and its chemical profiling by UPLC-Q-TOF-MS. BMC Complement. Altern. Med. 2018, 18, 54. [Google Scholar] [CrossRef]
- Qi, D.; Brownridge, P.; Xia, D.; Mackay, K.; Gonzalez-Galarza, F.F.; Kenyani, J.; Harman, V.; Beynon, R.J.; Jones, A.R. A Software Toolkit and Interface for Performing Stable Isotope Labeling and Top3 Quantification Using Progenesis LC-MS. OMICS A J. Integr. Biol. 2012, 16, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-Calling of Automated Sequencer Traces UsingPhred. I. Accuracy Assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res 2013, 2, 188. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Livak, K.; Schmittgen, T. Analysis of Relative Gene Expression Data using Real-Time Quantitative PCR. Methods 2002, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, S.; Zhao, C.; Ma, X.; Zhao, Y.; Shao, J.; Li, Y.; Li, H.; Song, H.; Ma, H.; et al. Transcriptional regulation of bark freezing tolerance in apple (Malus domestica Borkh.). Hortic. Res. 2020, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Pramsohler, M.; Hacker, J.; Neuner, G. Freezing pattern and frost killing temperature of apple (Malus domestica) wood under controlled conditions and in nature. Tree Physiol. 2012, 32, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, S.; Zhang, D.; Han, M.; Jin, X.; Zhao, C.; Wang, S.; Xing, L.; Ma, J.; Ji, J.; et al. Sequencing of a Wild Apple (Malus baccata) Genome Unravels the Differences Between Cultivated and Wild Apple Species Regarding Disease Resistance and Cold Tolerance. G3 Genes Genomes Genet. 2019, 9, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fu, X. Reprogramming of Plant Central Metabolism in Response to Abiotic Stresses: A Metabolomics View. Int. J. Mol. Sci. 2022, 23, 5716. [Google Scholar] [CrossRef]
- Sun, C.X.; Gao, X.X.; Li, M.Q.; Fu, J.Q.; Zhang, Y.L. Plastic responses in the metabolome and functional traits of maize plants to temperature variations. Plant Biol. 2016, 18, 249–261. [Google Scholar] [CrossRef]
- Ghassemi, S.; Delangiz, N.; Asgari Lajayer, B.; Saghafi, D.; Maggi, F. Review and future prospects on the mechanisms related to cold stress resistance and tolerance in medicinal plants. Acta Ecol. Sin. 2021, 41, 120–129. [Google Scholar] [CrossRef]
- Kaplan, F.; Kopka, J.; Haskell, D.W.; Zhao, W.; Schiller, K.C.; Gatzke, N.; Sung, D.Y.; Guy, C.L. Exploring the Temperature-Stress Metabolome of Arabidopsis. Plant Physiol. 2004, 136, 4159–4168. [Google Scholar] [CrossRef]
- Wienkoop, S.; Morgenthal, K.; Wolschin, F.; Scholz, M.; Selbig, J.; Weckwerth, W. Integration of Metabolomic and Proteomic Phenotypes: Analysis of Data Covariance Dissects Starch and RFO Metabolism from Low and High Temperature Compensation Response in Arabidopsis thaliana. Mol. Cell. Proteom. 2008, 7, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Mazzucotelli, E.; Tartari, A.; Cattivelli, L.; Forlani, G. Metabolism of γ-aminobutyric acid during cold acclimation and freezing and its relationship to frost tolerance in barley and wheat. J. Exp. Bot. 2006, 57, 3755–3766. [Google Scholar] [CrossRef]
- Korn, M.; Gärtner, T.; Erban, A.; Kopka, J.; Selbig, J.; Hincha, D.K. Predicting Arabidopsis Freezing Tolerance and Heterosis in Freezing Tolerance from Metabolite Composition. Mol. Plant 2010, 3, 224–235. [Google Scholar] [CrossRef]
- Wei, J.; Shen, Y.; Dong, X.; Zhu, Y.; Cui, J.; Li, H.; Zheng, G.; Tian, H.; Wang, Y.; Liu, Z. DNA methylation affects freezing tolerance in winter rapeseed by mediating the expression of genes related to JA and CK pathways. Front. Genet. 2022, 13, 968494. [Google Scholar] [CrossRef] [PubMed]
- Janmohammadi, M.; Zolla, L.; Rinalducci, S. Low temperature tolerance in plants: Changes at the protein level. Phytochemistry 2015, 117, 76–89. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Su, Y.; Dai, Z.W.; Lu, M.; Sun, W.; Yang, W.; Wu, S.S.; Wan, Z.T.; Wan, H.H.; Zhai, J. Integration of the metabolome and transcriptome reveals indigo biosynthesis in Phaius flavus flowers under freezing treatment. PeerJ 2022, 10, e13106. [Google Scholar] [CrossRef]
- Zheng, G.; Dong, X.; Wei, J.; Liu, Z.; Aslam, A.; Cui, J.; Li, H.; Wang, Y.; Tian, H.; Cao, X. Integrated methylome and transcriptome analysis unravel the cold tolerance mechanism in winter rapeseed(Brassica napus L.). BMC Plant Biol. 2022, 22, 414. [Google Scholar] [CrossRef]
- Liu, Q.; Kasuga, M.; Sakuma, Y.; Abe, H.; Miura, S.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Two Transcription Factors, DREB1 and DREB2, with an EREBP/AP2 DNA Binding Domain Separate Two Cellular Signal Transduction Pathways in Drought- and Low-Temperature-Responsive Gene Expression, Respectively, in Arabidopsis. Plant Cell 1998, 10, 1391–1406. [Google Scholar] [CrossRef]
- Raza, A.; Charagh, S.; Garcia-Caparros, P.; Rahman, M.A.; Ogwugwa, V.H.; Saeed, F.; Jin, W. Melatonin-mediated temperature stress tolerance in plants. GM Crops Food 2022, 13, 196–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ding, Y.; Shi, Y.; Ma, L.; Wang, Y.; Song, C.; Wilkins, K.A.; Davies, J.M.; Knight, H.; Knight, M.R.; et al. The calcium transporter ANNEXIN1 mediates cold-induced calcium signaling and freezing tolerance in plants. EMBO J. 2021, 40, e104559. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhao, Y.; Gu, J.; Zhou, M.; Gao, L.; Sun, R.-X.; Wang, W.-W.; Zhang, S.-H.; Yang, X.-J. Proteomic analysis reveals the molecular mechanism underlying the cold acclimation and freezing tolerance of wheat (Triticum aestivum L.). Plant Sci. 2022, 318, 111242. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Han, Z.; Chen, Y.; Huai, D.; Kang, Y.; Wang, Z.; Yan, L.; Jiang, H.; Lei, Y.; et al. Integrated Transcriptomics and Metabolomics Analysis Reveal Key Metabolism Pathways Contributing to Cold Tolerance in Peanut. Front. Plant Sci. 2021, 12, 752474. [Google Scholar] [CrossRef]
- Schulz, E.; Tohge, T.; Winkler, J.B.; Albert, A.; Schäffner, A.R.; Fernie, A.R.; Zuther, E.; Hincha, D.K. Natural Variation among Arabidopsis Accessions in the Regulation of Flavonoid Metabolism and Stress Gene Expression by Combined UV Radiation and Cold. Plant Cell Physiol. 2021, 62, 502–514. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Metabolites ID 1 | Gene ID 2 |
|---|---|---|
| Propanoate metabolism | pos_3699 | HF37733; HF39626 |
| Phenylalanine metabolism | pos_3975 | HF32403; NewGene_4237; NewGene_9458 |
| Fatty acid metabolism | pos_2511; pos_4795 | HF08643; HF37733; NewGene_7563 |
| Arginine and proline metabolism | pos_1927; pos_4010 | HF04894; HF27116; HF41275 |
| Carotenoid biosynthesis | pos_4978 | HF05780; HF05786; HF12739; HF23511; HF34033; HF06845 |
| Flavone and flavonol biosynthesis | pos_4009 | HF33398; HF33401 |
| Flavonoid biosynthesis | pos_3214 | HF00746; HF06720; HF07700; HF15427; HF21107; HF39985; HF42225 |
| Phenylpropanoid biosynthesis | pos_2056 | HF00746; HF06720; HF09713; HF16285; HF16814; HF29367; HF31060; HF39985; HF40459; NewGene_1559; NewGene_9073 |
| Steroid biosynthesis | pos_5705 | HF02832; HF16819; HF19558; HF20971; HF28860; HF28861; HF35942 |
| Histidine metabolism | pos_2179 | HF03481; HF29862 |
| Cyanoamino acid metabolism | pos_557 | HF32157; HF40459; HF44479 |
| Glycine, serine, and threonine metabolism | pos_1927; pos_557 | HF32157; HF32319; HF44479 |
| Tryptophan metabolism | pos_865 | HF09172; HF10553; HF10561; HF19130; HF22784; NewGene_6625; NewGene_9458 |
| Biosynthesis of amino acids | pos_557 | HF02826; HF02828; HF03481; HF04894; HF12391; HF27116; HF29862; HF32157; HF36209; HF38329; HF44479; NewGene_963 |
| Purine metabolism | pos_2642; pos_3608 | HF01558; HF04086; NewGene_495 |
| Tyrosine metabolism | pos_3561 | HF09884; HF32403; NewGene_4237; NewGene_9458 |
| Cutin, suberine, and wax biosynthesis | pos_4739; pos_4973 | HF05413; HF07533; HF22738; HF27176 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Wu, Y.; Liu, W.; Liu, J.; Li, P. Integration of Metabolome and Transcriptome Reveals the Major Metabolic Pathways and Potential Biomarkers in Response to Freeze-Stress Regulation in Apple (Malus domestica). Metabolites 2023, 13, 891. https://doi.org/10.3390/metabo13080891
Yu Y, Wu Y, Liu W, Liu J, Li P. Integration of Metabolome and Transcriptome Reveals the Major Metabolic Pathways and Potential Biomarkers in Response to Freeze-Stress Regulation in Apple (Malus domestica). Metabolites. 2023; 13(8):891. https://doi.org/10.3390/metabo13080891
Chicago/Turabian StyleYu, Yifei, YaJing Wu, Wenfei Liu, Jun Liu, and Ping Li. 2023. "Integration of Metabolome and Transcriptome Reveals the Major Metabolic Pathways and Potential Biomarkers in Response to Freeze-Stress Regulation in Apple (Malus domestica)" Metabolites 13, no. 8: 891. https://doi.org/10.3390/metabo13080891
APA StyleYu, Y., Wu, Y., Liu, W., Liu, J., & Li, P. (2023). Integration of Metabolome and Transcriptome Reveals the Major Metabolic Pathways and Potential Biomarkers in Response to Freeze-Stress Regulation in Apple (Malus domestica). Metabolites, 13(8), 891. https://doi.org/10.3390/metabo13080891