
Cross-Regulation of the Cellular Redox System, Oxygen, and Sphingolipid Signalling



Abstract
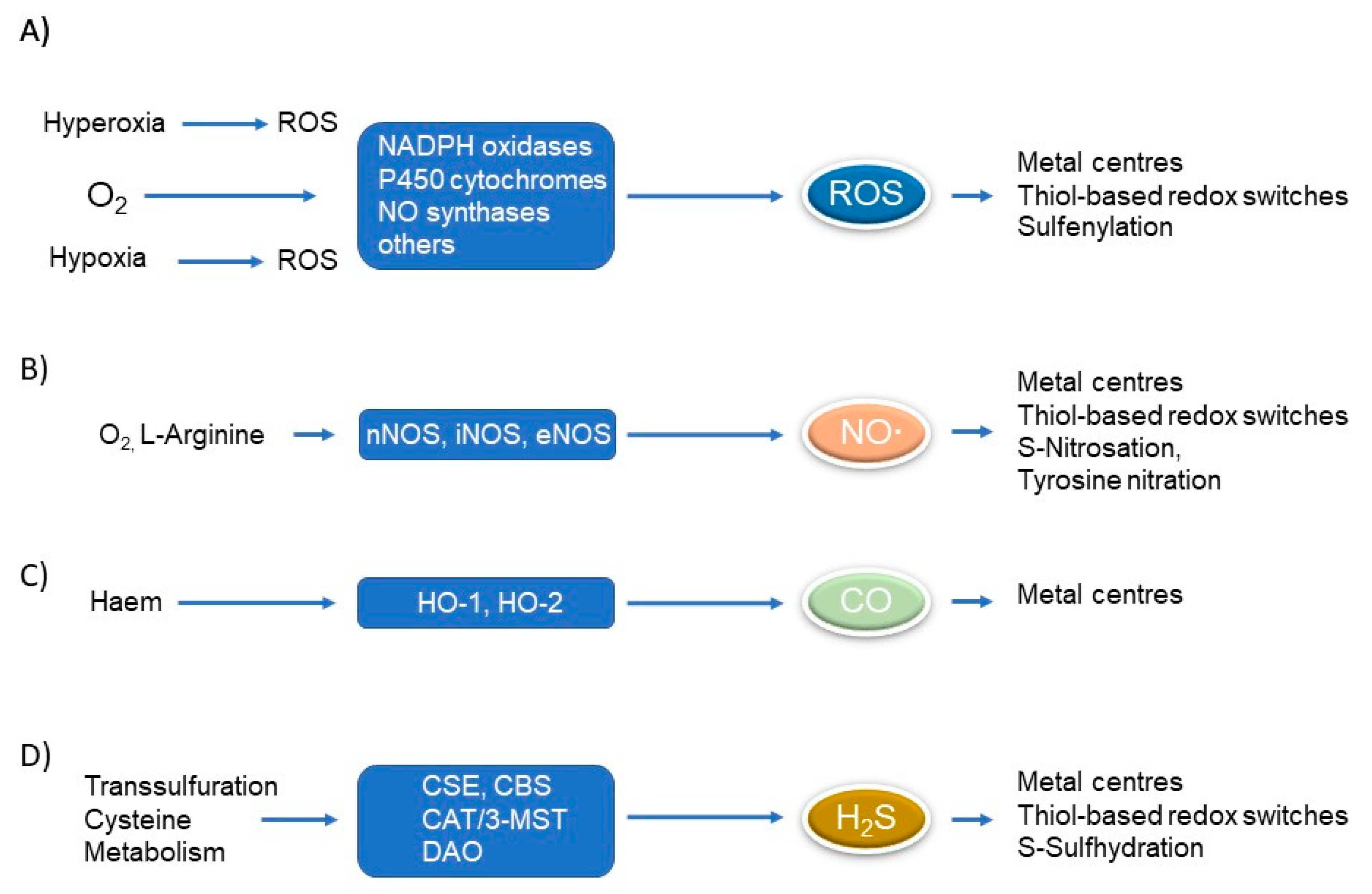
1. Introduction
1.1. Redox Signalling
1.2. Sphingolipid Biosynthesis, Degradation, and Signalling
2. Cross-Regulation of Redox and Sphingolipid Signalling
2.1. Hypoxia and Sphingolipid Signalling
2.2. Hyperoxia and Sphingolipid Signalling
2.3. ROS and Sphingolipid Signalling
2.4. Cross-Talk of Gasotransmitters and Sphingolipid Signalling
2.4.1. Nitric Oxide (NO∙)
2.4.2. Carbon Monoxide (CO)
2.4.3. Hydrogen Disulfide
3. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tse, H.M. The role of NADPH oxidases in infectious and inflammatory diseases. Redox Biol. 2021, 48, 102159. [Google Scholar] [CrossRef]
- Gabig, T.G.; Babior, B.M. The O2(-)-forming oxidase responsible for the respiratory burst in human neutrophils. Properties of the solubilized enzyme. J. Biol. Chem. 1979, 254, 9070–9074. [Google Scholar] [CrossRef]
- Sbarra, A.J.; Karnovsky, M.L. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J. Biol. Chem. 1959, 234, 1355–1362. [Google Scholar] [CrossRef]
- Forstermann, U. Janus-faced role of endothelial NO synthase in vascular disease: Uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol. Chem. 2006, 387, 1521–1533. [Google Scholar] [CrossRef]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Hibbs, J.B., Jr.; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric oxide: A cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94. [Google Scholar] [CrossRef]
- Marletta, M.A. Mammalian synthesis of nitrite, nitrate, nitric oxide, and N-nitrosating agents. Chem. Res. Toxicol. 1988, 1, 249–257. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Llewellyn-Smith, I.J.; Song, Z.M.; Costa, M.; Bredt, D.S.; Snyder, S.H. Ultrastructural localization of nitric oxide synthase immunoreactivity in guinea-pig enteric neurons. Brain Res. 1992, 577, 337–342. [Google Scholar] [CrossRef]
- Hevel, J.M.; White, K.A.; Marletta, M.A. Purification of the inducible murine macrophage nitric oxide synthase. Identification as a flavoprotein. J. Biol. Chem. 1991, 266, 22789–22791. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Cho, H.J.; Kwon, N.S.; Weise, M.F.; Nathan, C.F. Purification and characterization of the cytokine-induced macrophage nitric oxide synthase: An FAD- and FMN-containing flavoprotein. Proc. Natl. Acad. Sci. USA 1991, 88, 7773–7777. [Google Scholar] [CrossRef]
- Pfeilschifter, J.; Schwarzenbach, H. Interleukin 1 and tumor necrosis factor stimulate cGMP formation in rat renal mesangial cells. FEBS Lett. 1990, 273, 185–187. [Google Scholar] [CrossRef]
- Busse, R.; Mulsch, A. Induction of nitric oxide synthase by cytokines in vascular smooth muscle cells. FEBS Lett. 1990, 275, 87–90. [Google Scholar] [CrossRef]
- Bender, D.; Schwarz, G. Nitrite-dependent nitric oxide synthesis by molybdenum enzymes. FEBS Lett. 2018, 592, 2126–2139. [Google Scholar] [CrossRef]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef]
- Williams, S.E.; Wootton, P.; Mason, H.S.; Bould, J.; Iles, D.E.; Riccardi, D.; Peers, C.; Kemp, P.J. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science 2004, 306, 2093–2097. [Google Scholar] [CrossRef]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Kimura, H. Signaling of hydrogen sulfide and polysulfides. Antioxid. Redox Signal. 2015, 22, 347–349. [Google Scholar] [CrossRef]
- Beck, K.F.; Pfeilschifter, J. The Pathophysiology of H2S in Renal Glomerular Diseases. Biomolecules 2022, 12, 207. [Google Scholar] [CrossRef]
- Wang, M.; Guo, Z.; Wang, S. The binding site for the transcription factor, NF-kappaB, on the cystathionine gamma-lyase promoter is critical for LPS-induced cystathionine gamma-lyase expression. Int. J. Mol. Med. 2014, 34, 639–645. [Google Scholar] [CrossRef]
- Hassan, M.I.; Boosen, M.; Schaefer, L.; Kozlowska, J.; Eisel, F.; von Knethen, A.; Beck, M.; Hemeida, R.A.; El-Moselhy, M.A.; Hamada, F.M.; et al. Platelet-derived growth factor-BB induces cystathionine gamma-lyase expression in rat mesangial cells via a redox-dependent mechanism. Br. J. Pharmacol. 2012, 166, 2231–2242. [Google Scholar] [CrossRef]
- Beck, K.F.; Pfeilschifter, J. Gasotransmitter synthesis and signalling in the renal glomerulus. Implications for glomerular diseases. Cell. Signal. 2021, 77, 109823. [Google Scholar] [CrossRef]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Han, G.; Gupta, S.D.; Gable, K.; Niranjanakumari, S.; Moitra, P.; Eichler, F.; Brown, R.H., Jr.; Harmon, J.M.; Dunn, T.M. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc. Natl. Acad. Sci. USA 2009, 106, 8186–8191. [Google Scholar] [CrossRef]
- Glueck, M.; Koch, A.; Brunkhorst, R.; Bouzas, N.F.; Trautmann, S.; Schaefer, L.; Pfeilschifter, W.; Pfeilschifter, J.; Vutukuri, R. The atypical sphingosine 1-phosphate variant, d16:1 S1P, mediates CTGF induction via S1P2 activation in renal cell carcinoma. FEBS J. 2022, 289, 5670–5681. [Google Scholar] [CrossRef]
- Davis, D.; Kannan, M.; Wattenberg, B. Orm/ORMDL proteins: Gate guardians and master regulators. Adv. Biol. Regul. 2018, 70, 3–18. [Google Scholar] [CrossRef]
- Gupta, S.D.; Gable, K.; Han, G.; Borovitskaya, A.; Selby, L.; Dunn, T.M.; Harmon, J.M. Tsc10p and FVT1: Topologically distinct short-chain reductases required for long-chain base synthesis in yeast and mammals. J. Lipid Res. 2009, 50, 1630–1640. [Google Scholar] [CrossRef]
- Park, J.W.; Park, W.J.; Futerman, A.H. Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochim. Biophys. Acta 2014, 1841, 671–681. [Google Scholar] [CrossRef]
- Michel, C.; van Echten-Deckert, G.; Rother, J.; Sandhoff, K.; Wang, E.; Merrill, A.H., Jr. Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide. J. Biol. Chem. 1997, 272, 22432–22437. [Google Scholar] [CrossRef]
- Kumagai, K.; Hanada, K. Structure, functions and regulation of CERT, a lipid-transfer protein for the delivery of ceramide at the ER-Golgi membrane contact sites. FEBS Lett. 2019, 593, 2366–2377. [Google Scholar] [CrossRef]
- Dunn, T.M.; Tifft, C.J.; Proia, R.L. A perilous path: The inborn errors of sphingolipid metabolism. J. Lipid Res. 2019, 60, 475–483. [Google Scholar] [CrossRef]
- Sandhoff, R.; Schulze, H.; Sandhoff, K. Ganglioside Metabolism in Health and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 1–62. [Google Scholar]
- Alemany, R.; van Koppen, C.J.; Danneberg, K.; Ter Braak, M.; Meyer Zu Heringdorf, D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 374, 413–428. [Google Scholar] [CrossRef]
- Adams, D.R.; Pyne, S.; Pyne, N.J. Sphingosine Kinases: Emerging Structure-Function Insights. Trends Biochem. Sci. 2016, 41, 395–409. [Google Scholar] [CrossRef]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef]
- Saba, J.D. Fifty years of lyase and a moment of truth: Sphingosine phosphate lyase from discovery to disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J. New players on the center stage: Sphingosine 1-phosphate and its receptors as drug targets. Biochem. Pharmacol. 2008, 75, 1893–1900. [Google Scholar] [CrossRef]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Investig. 2015, 125, 1379–1387. [Google Scholar] [CrossRef]
- Yee Koh, M.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef]
- Caro, J. Hypoxia regulation of gene transcription. High Alt. Med. Biol. 2001, 2, 145–154. [Google Scholar] [CrossRef]
- Kennel, K.B.; Burmeister, J.; Schneider, M.; Taylor, C.T. The PHD1 oxygen sensor in health and disease. J. Physiol. 2018, 596, 3899–3913. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Kendler, A.; Dawson, G. Progressive hypoxia inhibits the de novo synthesis of galactosylceramide in cultured oligodendrocytes. J. Biol. Chem. 1990, 265, 12259–12266. [Google Scholar] [CrossRef]
- Hernandez, O.M.; Discher, D.J.; Bishopric, N.H.; Webster, K.A. Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ. Res. 2000, 86, 198–204. [Google Scholar] [CrossRef]
- Zhu, Q.; Lin, L.; Cheng, Q.; Xu, Q.; Zhang, J.; Tomlinson, S.; Jin, J.; Chen, X.; He, S. The role of acid sphingomyelinase and caspase 5 in hypoxia-induced HuR cleavage and subsequent apoptosis in hepatocytes. Biochim. Biophys. Acta 2012, 1821, 1453–1461. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Frazziano, G.; Moral-Sanz, J.; Menendez, C.; Castaneda, J.; Gonzalez, C.; Villamor, E.; Perez-Vizcaino, F. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 2009, 82, 296–302. [Google Scholar] [CrossRef]
- Yun, J.K.; Kester, M. Regulatory role of sphingomyelin metabolites in hypoxia-induced vascular smooth muscle cell proliferation. Arch. Biochem. Biophys. 2002, 408, 78–86. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Wang, P.; Zhang, S.Y.; Dong, Y.; Zeng, G.; Yan, Y.; Sun, L.; Wu, Q.; Liu, H.; et al. Adipocyte Hypoxia-Inducible Factor 2alpha Suppresses Atherosclerosis by Promoting Adipose Ceramide Catabolism. Cell Metab. 2019, 30, 937–951.e5. [Google Scholar] [CrossRef]
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. J. Biol. Chem. 2008, 283, 3365–3375. [Google Scholar] [CrossRef]
- Schwalm, S.; Doll, F.; Romer, I.; Bubnova, S.; Pfeilschifter, J.; Huwiler, A. Sphingosine kinase-1 is a hypoxia-regulated gene that stimulates migration of human endothelial cells. Biochem. Biophys. Res. Commun. 2008, 368, 1020–1025. [Google Scholar] [CrossRef]
- Ahmad, M.; Long, J.S.; Pyne, N.J.; Pyne, S. The effect of hypoxia on lipid phosphate receptor and sphingosine kinase expression and mitogen-activated protein kinase signaling in human pulmonary smooth muscle cells. Prostaglandins Other Lipid Mediat. 2006, 79, 278–286. [Google Scholar] [CrossRef]
- Schnitzer, S.E.; Welgert, A.; Zhou, J.; Brune, B. Hypoxia Enhances Sphingosine Kinase 2 Activity and Provokes Sphingosine-1-Phosphate-Mediated Chemoresistance in A549 Lung Cancer Cells. Mol. Cancer Res. 2009, 7, 393–401. [Google Scholar] [CrossRef]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef]
- Hait, N.C.; Maiti, A.; Xu, P.; Qi, Q.; Kawaguchi, T.; Okano, M.; Takabe, K.; Yan, L.; Luo, C. Regulation of hypoxia-inducible factor functions in the nucleus by sphingosine-1-phosphate. FASEB J. 2020, 34, 4293–4310. [Google Scholar] [CrossRef]
- Li, C.; Wu, B.; Li, Y.; Liu, Y.; Wang, J.; Xie, J.; Xu, X.; Tian, X.; Ye, Z.; Guan, J.; et al. Loss of sphingosine kinase 2 promotes the expansion of hematopoietic stem cells by improving their metabolic fitness. Blood 2022, 140, 1686–1701. [Google Scholar] [CrossRef]
- Bouquerel, P.; Gstalder, C.; Muller, D.; Laurent, J.; Brizuela, L.; Sabbadini, R.A.; Malavaud, B.; Pyronnet, S.; Martineau, Y.; Ader, I.; et al. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2alpha expression and activity in cancer. Oncogenesis 2016, 5, e209. [Google Scholar] [CrossRef]
- Rosenberger, C.; Mandriota, S.; Jurgensen, J.S.; Wiesener, M.S.; Horstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.U. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732. [Google Scholar] [CrossRef]
- Hafizi, R.; Imeri, F.; Stepanovska Tanturovska, B.; Manaila, R.; Schwalm, S.; Trautmann, S.; Wenger, R.H.; Pfeilschifter, J.; Huwiler, A. Sphk1 and Sphk2 Differentially Regulate Erythropoietin Synthesis in Mouse Renal Interstitial Fibroblast-like Cells. Int. J. Mol. Sci. 2022, 23, 5882. [Google Scholar] [CrossRef]
- Gaspersic, J.; Kristan, A.; Kunej, T.; Zupan, I.P.; Debeljak, N. Erythrocytosis: Genes and pathways involved in disease development. Blood Transfus. 2021, 19, 518–532. [Google Scholar]
- Kang, M.S.; Ahn, K.H.; Kim, S.K.; Jeon, H.J.; Ji, J.E.; Choi, J.M.; Jung, K.M.; Jung, S.Y.; Kim, D.K. Hypoxia-induced neuronal apoptosis is mediated by de novo synthesis of ceramide through activation of serine palmitoyltransferase. Cell. Signal. 2010, 22, 610–618. [Google Scholar] [CrossRef]
- Takagi, S.; Tojo, H.; Tomita, S.; Sano, S.; Itami, S.; Hara, M.; Inoue, S.; Horie, K.; Kondoh, G.; Hosokawa, K.; et al. Alteration of the 4-sphingenine scaffolds of ceramides in keratinocyte-specific Arnt-deficient mice affects skin barrier function. J. Clin. Investig. 2003, 112, 1372–1382. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. The role of sphingolipid metabolism in cutaneous permeability barrier formation. Biochim. Biophys. Acta 2014, 1841, 441–452. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Domann, F.E. Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1alpha signaling node. Chem. Biol. Interact. 2014, 218, 82–88. [Google Scholar] [CrossRef]
- Takacova, M.; Holotnakova, T.; Vondracek, J.; Machala, M.; Pencikova, K.; Gradin, K.; Poellinger, L.; Pastorek, J.; Pastorekova, S.; Kopacek, J. Role of aryl hydrocarbon receptor in modulation of the expression of the hypoxia marker carbonic anhydrase IX. Biochem. J. 2009, 419, 419–425. [Google Scholar] [CrossRef]
- Majumder, S.; Kono, M.; Lee, Y.T.; Byrnes, C.; Li, C.; Tuymetova, G.; Proia, R.L. A genome-wide CRISPR/Cas9 screen reveals that the aryl hydrocarbon receptor stimulates sphingolipid levels. J. Biol. Chem. 2020, 295, 4341–4349. [Google Scholar] [CrossRef]
- Juricek, L.; Carcaud, J.; Pelhaitre, A.; Riday, T.T.; Chevallier, A.; Lanzini, J.; Auzeil, N.; Laprevote, O.; Dumont, F.; Jacques, S.; et al. AhR-deficiency as a cause of demyelinating disease and inflammation. Sci. Rep. 2017, 7, 9794. [Google Scholar] [CrossRef]
- Burtscher, J.; Mallet, R.T.; Pialoux, V.; Millet, G.P.; Burtscher, M. Adaptive Responses to Hypoxia and/or Hyperoxia in Humans. Antioxid. Redox Signal. 2022, 37, 887–912. [Google Scholar] [CrossRef]
- Harijith, A.; Pendyala, S.; Reddy, N.M.; Bai, T.; Usatyuk, P.V.; Berdyshev, E.; Gorshkova, I.; Huang, L.S.; Mohan, V.; Garzon, S.; et al. Sphingosine kinase 1 deficiency confers protection against hyperoxia-induced bronchopulmonary dysplasia in a murine model: Role of S1P signaling and Nox proteins. Am. J. Pathol. 2013, 183, 1169–1182. [Google Scholar] [CrossRef]
- Ha, A.W.; Sudhadevi, T.; Ebenezer, D.L.; Fu, P.; Berdyshev, E.V.; Ackerman, S.J.; Natarajan, V.; Harijith, A. Neonatal therapy with PF543, a sphingosine kinase 1 inhibitor, ameliorates hyperoxia-induced airway remodeling in a murine model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L497–L512. [Google Scholar] [CrossRef]
- Harijith, A.; Pendyala, S.; Ebenezer, D.L.; Ha, A.W.; Fu, P.; Wang, Y.T.; Ma, K.; Toth, P.T.; Berdyshev, E.V.; Kanteti, P.; et al. Hyperoxia-induced p47phox activation and ROS generation is mediated through S1P transporter Spns2, and S1P/S1P1&2 signaling axis in lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L337–L351. [Google Scholar]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef]
- Liu, B.; Hannun, Y.A. Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione. J. Biol. Chem. 1997, 272, 16281–16287. [Google Scholar] [CrossRef]
- Mansat-de Mas, V.; Bezombes, C.; Quillet-Mary, A.; Bettaieb, A.; D’Orgeix, A.D.; Laurent, G.; Jaffrezou, J.P. Implication of radical oxygen species in ceramide generation, c-Jun N-terminal kinase activation and apoptosis induced by daunorubicin. Mol. Pharmacol. 1999, 56, 867–874. [Google Scholar]
- Won, J.S.; Singh, I. Sphingolipid signaling and redox regulation. Free Radic. Biol. Med. 2006, 40, 1875–1888. [Google Scholar] [CrossRef]
- Li, P.L.; Gulbins, E. Bioactive Lipids and Redox Signaling: Molecular Mechanism and Disease Pathogenesis. Antioxid. Redox Signal. 2018, 28, 911–915. [Google Scholar] [CrossRef]
- Bhat, O.M.; Yuan, X.; Li, G.; Lee, R.; Li, P.L. Sphingolipids and Redox Signaling in Renal Regulation and Chronic Kidney Diseases. Antioxid. Redox Signal. 2018, 28, 1008–1026. [Google Scholar] [CrossRef]
- Ueda, N. A Rheostat of Ceramide and Sphingosine-1-Phosphate as a Determinant of Oxidative Stress-Mediated Kidney Injury. Int. J. Mol. Sci. 2022, 23, 4010. [Google Scholar] [CrossRef]
- Singh, I.; Pahan, K.; Khan, M.; Singh, A.K. Cytokine-mediated induction of ceramide production is redox-sensitive. Implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J. Biol. Chem. 1998, 273, 20354–20362. [Google Scholar] [CrossRef]
- Gouaze, V.; Mirault, M.E.; Carpentier, S.; Salvayre, R.; Levade, T.; Andrieu-Abadie, N. Glutathione peroxidase-1 overexpression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol. Pharmacol. 2001, 60, 488–496. [Google Scholar]
- Huwiler, A.; Boddinghaus, B.; Pautz, A.; Dorsch, S.; Franzen, R.; Briner, V.A.; Brade, V.; Pfeilschifter, J. Superoxide potently induces ceramide formation in glomerular endothelial cells. Biochem. Biophys. Res. Commun. 2001, 284, 404–410. [Google Scholar] [CrossRef]
- Pautz, A.; Franzen, R.; Dorsch, S.; Boddinghaus, B.; Briner, V.A.; Pfeilschifter, J.; Huwiler, A. Cross-talk between nitric oxide and superoxide determines ceramide formation and apoptosis in glomerular cells. Kidney Int. 2002, 61, 790–796. [Google Scholar] [CrossRef]
- Xiang, H.; Jin, S.; Tan, F.; Xu, Y.; Lu, Y.; Wu, T. Physiological functions and therapeutic applications of neutral sphingomyelinase and acid sphingomyelinase. Biomed. Pharmacother. 2021, 139, 111610. [Google Scholar] [CrossRef]
- Wu, B.X.; Clarke, C.J.; Hannun, Y.A. Mammalian neutral sphingomyelinases: Regulation and roles in cell signaling responses. Neuromol. Med. 2010, 12, 320–330. [Google Scholar] [CrossRef]
- Ratnayake, S.; Dias, I.H.; Lattman, E.; Griffiths, H.R. Stabilising cysteinyl thiol oxidation and nitrosation for proteomic analysis. J. Proteom. 2013, 92, 160–170. [Google Scholar] [CrossRef]
- Rodrigues-Lima, F.; Fensome, A.C.; Josephs, M.; Evans, J.; Veldman, R.J.; Katan, M. Structural requirements for catalysis and membrane targeting of mammalian enzymes with neutral sphingomyelinase and lysophospholipid phospholipase C activities. Analysis by chemical modification and site-directed mutagenesis. J. Biol. Chem. 2000, 275, 28316–28325. [Google Scholar] [CrossRef]
- Fensome, A.C.; Rodrigues-Lima, F.; Josephs, M.; Paterson, H.F.; Katan, M. A neutral magnesium-dependent sphingomyelinase isoform associated with intracellular membranes and reversibly inhibited by reactive oxygen species. J. Biol. Chem. 2000, 275, 1128–1136. [Google Scholar] [CrossRef]
- Josephs, M.; Katan, M.; Rodrigues-Lima, F. Irreversible inactivation of magnesium-dependent neutral sphingomyelinase 1 (NSM1) by peroxynitrite, a nitric oxide-derived oxidant. FEBS Lett. 2002, 531, 329–334. [Google Scholar] [CrossRef]
- Dotson, P.P., 2nd; Karakashian, A.A.; Nikolova-Karakashian, M.N. Neutral sphingomyelinase-2 is a redox sensitive enzyme: Role of catalytic cysteine residues in regulation of enzymatic activity through changes in oligomeric state. Biochem. J. 2015, 465, 371–382. [Google Scholar] [CrossRef]
- Bernardo, K.; Krut, O.; Wiegmann, K.; Kreder, D.; Micheli, M.; Schafer, R.; Sickman, A.; Schmidt, W.E.; Schroder, J.M.; Meyer, H.E.; et al. Purification and characterization of a magnesium-dependent neutral sphingomyelinase from bovine brain. J. Biol. Chem. 2000, 275, 7641–7647. [Google Scholar] [CrossRef]
- Krut, O.; Wiegmann, K.; Kashkar, H.; Yazdanpanah, B.; Kronke, M. Novel tumor necrosis factor-responsive mammalian neutral sphingomyelinase-3 is a C-tail-anchored protein. J. Biol. Chem. 2006, 281, 13784–13793. [Google Scholar] [CrossRef]
- Moylan, J.S.; Smith, J.D.; Wolf Horrell, E.M.; McLean, J.B.; Deevska, G.M.; Bonnell, M.R.; Nikolova-Karakashian, M.N.; Reid, M.B. Neutral sphingomyelinase-3 mediates TNF-stimulated oxidant activity in skeletal muscle. Redox Biol. 2014, 2, 910–920. [Google Scholar] [CrossRef]
- Qiu, H.; Edmunds, T.; Baker-Malcolm, J.; Karey, K.P.; Estes, S.; Schwarz, C.; Hughes, H.; Van Patten, S.M. Activation of human acid sphingomyelinase through modification or deletion of C-terminal cysteine. J. Biol. Chem. 2003, 278, 32744–32752. [Google Scholar] [CrossRef]
- He, X.; Miranda, S.R.; Xiong, X.; Dagan, A.; Gatt, S.; Schuchman, E.H. Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim. Biophys. Acta 1999, 1432, 251–264. [Google Scholar] [CrossRef]
- Gudz, T.I.; Tserng, K.Y.; Hoppel, C.L. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 1997, 272, 24154–24158. [Google Scholar] [CrossRef]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Chenna, S.; Koopman, W.J.H.; Prehn, J.H.M.; Connolly, N.M.C. Mechanisms and mathematical modeling of ROS production by the mitochondrial electron transport chain. Am. J. Physiol. Cell Physiol. 2022, 323, C69–C83. [Google Scholar] [CrossRef]
- Siskind, L.J.; Colombini, M. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J. Biol. Chem. 2000, 275, 38640–38644. [Google Scholar] [CrossRef]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 2002, 277, 26796–26803. [Google Scholar] [CrossRef]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Muller, D.; Schneider, M.; et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Lauterwasser, J.; Todt, F.; Zerbes, R.M.; Nguyen, T.N.; Craigen, W.; Lazarou, M.; van der Laan, M.; Edlich, F. The porin VDAC2 is the mitochondrial platform for Bax retrotranslocation. Sci. Rep. 2016, 6, 32994. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Kleuser, B.; Huwiler, A.; Arenz, C. Effective inhibition of acid and neutral ceramidases by novel B-13 and LCL-464 analogues. Bioorg. Med. Chem. 2013, 21, 874–882. [Google Scholar] [CrossRef]
- Flowers, M.; Fabrias, G.; Delgado, A.; Casas, J.; Abad, J.L.; Cabot, M.C. C6-ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res. Treat. 2012, 133, 447–458. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Jin, Y.; Wang; He, Q.; Liu, Z.; Ai, Q.; Lei, Y.; Li, Y.; Song, F.; et al. Alkaline ceramidase 2 is a novel direct target of p53 and induces autophagy and apoptosis through ROS generation. Sci. Rep. 2017, 7, 44573. [Google Scholar] [CrossRef]
- Taniai, T.; Shirai, Y.; Shimada, Y.; Hamura, R.; Yanagaki, M.; Takada, N.; Horiuchi, T.; Haruki, K.; Furukawa, K.; Uwagawa, T.; et al. Inhibition of acid ceramidase elicits mitochondrial dysfunction and oxidative stress in pancreatic cancer cells. Cancer Sci. 2021, 112, 4570–4579. [Google Scholar] [CrossRef]
- Malvi, P.; Janostiak, R.; Nagarajan, A.; Zhang, X.; Wajapeyee, N. N-acylsphingosine amidohydrolase 1 promotes melanoma growth and metastasis by suppressing peroxisome biogenesis-induced ROS production. Mol. Metab. 2021, 48, 101217. [Google Scholar] [CrossRef]
- Li, Q.; Qian, J.; Li, Y.; Huang, P.; Liang, H.; Sun, H.; Liu, C.; Peng, J.; Lin, X.; Chen, X.; et al. Generation of sphingosine-1-phosphate by sphingosine kinase 1 protects nonalcoholic fatty liver from ischemia/reperfusion injury through alleviating reactive oxygen species production in hepatocytes. Free Radic. Biol. Med. 2020, 159, 136–149. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Schuchman, E.H. Ceramide and Ischemia/Reperfusion Injury. J. Lipids 2018, 2018, 3646725. [Google Scholar] [CrossRef]
- Shao, J.J.; Peng, Y.; Wang, L.M.; Wang, J.K.; Chen, X. Activation of SphK1 by K6PC-5 Inhibits Oxygen-Glucose Deprivation/Reoxygenation-Induced Myocardial Cell Death. DNA Cell Biol. 2015, 34, 669–676. [Google Scholar] [CrossRef]
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49. [Google Scholar] [CrossRef]
- Ren, S.; Xin, C.; Pfeilschifter, J.; Huwiler, A. A novel mode of action of the putative sphingosine kinase inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SKI II): Induction of lysosomal sphingosine kinase 1 degradation. Cell Physiol. Biochem. 2010, 26, 97–104. [Google Scholar] [CrossRef]
- Loveridge, C.; Tonelli, F.; Leclercq, T.; Lim, K.G.; Long, J.S.; Berdyshev, E.; Tate, R.J.; Natarajan, V.; Pitson, S.M.; Pyne, N.J.; et al. The sphingosine kinase 1 inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl)thiazole induces proteasomal degradation of sphingosine kinase 1 in mammalian cells. J. Biol. Chem. 2010, 285, 38841–38852. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Lidington, D.; Vogel, L.; Peter, B.F.; Sohn, H.Y.; Pagano, P.J.; Pitson, S.; Spiegel, S.; Pohl, U.; Bolz, S.S. Sphingosine kinase functionally links elevated transmural pressure and increased reactive oxygen species formation in resistance arteries. FASEB J. 2006, 20, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Ohkura, S.; Takashima, S.; Ohtani, K.; Okamoto, Y.; Tanaka, T.; Hirano, K.; Usui, S.; Wang, F.; Du, W.; et al. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc. Res. 2010, 85, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Pfeilschifter, J. Nitric oxide signalling with a special focus on lipid-derived mediators. Biol. Chem. 2003, 384, 1379–1389. [Google Scholar] [CrossRef]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef]
- Gruetter, C.A.; Barry, B.K.; McNamara, D.B.; Gruetter, D.Y.; Kadowitz, P.J.; Ignarro, L. Relaxation of bovine coronary artery and activation of coronary arterial guanylate cyclase by nitric oxide, nitroprusside and a carcinogenic nitrosoamine. J. Cycl. Nucleotide Res. 1979, 5, 211–224. [Google Scholar]
- Sharma, V.; Fernando, V.; Letson, J.; Walia, Y.; Zheng, X.; Fackelman, D.; Furuta, S. S-Nitrosylation in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4600. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res. 2020, 45, 2815–2827. [Google Scholar] [CrossRef]
- Zhan, X.; Desiderio, D.M. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006, 354, 279–289. [Google Scholar] [CrossRef]
- Lau, B.; Fazelinia, H.; Mohanty, I.; Raimo, S.; Tenopoulou, M.; Doulias, P.T.; Ischiropoulos, H. Endogenous S-nitrosocysteine proteomic inventories identify a core of proteins in heart metabolic pathways. Redox Biol. 2021, 47, 102153. [Google Scholar] [CrossRef] [PubMed]
- Lander, H.M.; Ogiste, J.S.; Pearce, S.F.; Levi, R.; Novogrodsky, A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J. Biol. Chem. 1995, 270, 7017–7020. [Google Scholar] [CrossRef] [PubMed]
- Pfeilschifter, J.; Huwiler, A. Nitric oxide stimulates stress-activated protein kinases in glomerular endothelial and mesangial cells. FEBS Lett. 1996, 396, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Callsen, D.; Pfeilschifter, J.; Brune, B. Rapid and delayed p42/p44 mitogen-activated protein kinase activation by nitric oxide: The role of cyclic GMP and tyrosine phosphatase inhibition. J. Immunol. 1998, 161, 4852–4858. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J. Nitric oxide stimulates the stress-activated protein kinase p38 in rat renal mesangial cells. J. Exp. Biol. 1999, 202 Pt 6, 655–660. [Google Scholar] [CrossRef]
- Lander, H.M.; Jacovina, A.T.; Davis, R.J.; Tauras, J.M. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J. Biol. Chem. 1996, 271, 19705–19709. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J.; van den Bosch, H. Nitric oxide donors induce stress signaling via ceramide formation in rat renal mesangial cells. J. Biol. Chem. 1999, 274, 7190–7195. [Google Scholar] [CrossRef]
- Huwiler, A.; Dorsch, S.; Briner, V.A.; van den Bosch, H.; Pfeilschifter, J. Nitric oxide stimulates chronic ceramide formation in glomerular endothelial cells. Biochem. Biophys. Res. Commun. 1999, 258, 60–65. [Google Scholar] [CrossRef]
- Falcone, S.; Perrotta, C.; De Palma, C.; Pisconti, A.; Sciorati, C.; Capobianco, A.; Rovere-Querini, P.; Manfredi, A.A.; Clementi, E. Activation of acid sphingomyelinase and its inhibition by the nitric oxide/cyclic guanosine 3′,5′-monophosphate pathway: Key events in Escherichia coli-elicited apoptosis of dendritic cells. J. Immunol. 2004, 173, 4452–4463. [Google Scholar] [CrossRef]
- Barsacchi, R.; Perrotta, C.; Sestili, P.; Cantoni, O.; Moncada, S.; Clementi, E. Cyclic GMP-dependent inhibition of acid sphingomyelinase by nitric oxide: An early step in protection against apoptosis. Cell Death Differ. 2002, 9, 1248–1255. [Google Scholar] [CrossRef]
- De Nadai, C.; Sestili, P.; Cantoni, O.; Lievremont, J.P.; Sciorati, C.; Barsacchi, R.; Moncada, S.; Meldolesi, J.; Clementi, E. Nitric oxide inhibits tumor necrosis factor-alpha-induced apoptosis by reducing the generation of ceramide. Proc. Natl. Acad. Sci. USA 2000, 97, 5480–5485. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated with Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9, 1186. [Google Scholar] [CrossRef]
- Schlossmann, J.; Desch, M. cGK substrates. Handb. Exp. Pharmacol. 2009, 191, 163–193. [Google Scholar]
- Matsumoto, A.; Comatas, K.E.; Liu, L.; Stamler, J.S. Screening for nitric oxide-dependent protein-protein interactions. Science 2003, 301, 657–661. [Google Scholar] [CrossRef]
- Franzen, R.; Pautz, A.; Brautigam, L.; Geisslinger, G.; Pfeilschifter, J.; Huwiler, A. Interleukin-1beta induces chronic activation and de novo synthesis of neutral ceramidase in renal mesangial cells. J. Biol. Chem. 2001, 276, 35382–35389. [Google Scholar] [CrossRef] [PubMed]
- Franzen, R.; Fabbro, D.; Aschrafi, A.; Pfeilschifter, J.; Huwiler, A. Nitric oxide induces degradation of the neutral ceramidase in rat renal mesangial cells and is counterregulated by protein kinase C. J. Biol. Chem. 2002, 277, 46184–46190. [Google Scholar] [CrossRef] [PubMed]
- Franzen, R.; Pfeilschifter, J.; Huwiler, A. Nitric oxide induces neutral ceramidase degradation by the ubiquitin/proteasome complex in renal mesangial cell cultures. FEBS Lett. 2002, 532, 441–444. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Carbon monoxide: Present and future indications for a medical gas. Korean J. Intern. Med. 2013, 28, 123–140. [Google Scholar] [CrossRef]
- Piantadosi, C.A. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic. Biol. Med. 2008, 45, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Adach, W.; Olas, B. Carbon monoxide and its donors—Their implications for medicine. Future Med. Chem. 2019, 11, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Ryter, S.W.; Choi, A.M. CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 411–449. [Google Scholar] [CrossRef]
- Brune, B.; Ullrich, V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol. Pharmacol. 1987, 32, 497–504. [Google Scholar] [PubMed]
- Arnold, W.P.; Aldred, R.; Murad, F. Cigarette smoke activates guanylate cyclase and increases guanosine 3′,5′-monophosphate in tissues. Science 1977, 198, 934–936. [Google Scholar] [CrossRef] [PubMed]
- Graser, T.; Vedernikov, Y.P.; Li, D.S. Study on the mechanism of carbon monoxide induced endothelium-independent relaxation in porcine coronary artery and vein. Biomed. Biochim. Acta 1990, 49, 293–296. [Google Scholar]
- Wender, M. Studies of Cerebral Lipids in a Relapsing Case of Carbon Monoxide Poisoning. Acta Neuropathol. 1963, 3, 371–377. [Google Scholar] [CrossRef]
- Mawatari, S. Biochemical study on rat brain in acute carbon monoxide poisoning. Folia Psychiatr. Neurol. Jpn. 1970, 24, 123–129. [Google Scholar] [CrossRef]
- Weis, N.; Weigert, A.; von Knethen, A.; Brune, B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol. Biol. Cell 2009, 20, 1280–1288. [Google Scholar] [CrossRef]
- Abdelbaset-Ismail, A.; Cymer, M.; Borkowska-Rzeszotek, S.; Brzezniakiewicz-Janus, K.; Rameshwar, P.; Kakar, S.S.; Ratajczak, J.; Ratajczak, M.Z. Bioactive Phospholipids Enhance Migration and Adhesion of Human Leukemic Cells by Inhibiting Heme Oxygenase 1 (HO-1) and Inducible Nitric Oxygenase Synthase (iNOS) in a p38 MAPK-Dependent Manner. Stem Cell Rev. Rep. 2019, 15, 139–154. [Google Scholar] [CrossRef]
- Jung, J.S.; Choi, M.J.; Ko, H.M.; Kim, H.S. Short-chain C2 ceramide induces heme oxygenase-1 expression by upregulating AMPK and MAPK signaling pathways in rat primary astrocytes. Neurochem. Int. 2016, 94, 39–47. [Google Scholar] [CrossRef]
- Mitidieri, E.; Gurgone, D.; Caiazzo, E.; Tramontano, T.; Cicala, C.; Sorrentino, R.; d’Emmanuele di Villa Bianca, R. L-cysteine/cystathionine-beta-synthase-induced relaxation in mouse aorta involves a L-serine/sphingosine-1-phosphate/NO pathway. Br. J. Pharmacol. 2020, 177, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell Longev. 2016, 2016, 6904327. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Moore, P.K. Hydrogen sulfide and the vasculature: A novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell Mol. Med. 2009, 13, 488–507. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Kumazoe, M.; Yamashita, S.; Tachibana, H. Hydrogen sulphide donors selectively potentiate a green tea polyphenol EGCG-induced apoptosis of multiple myeloma cells. Sci. Rep. 2017, 7, 6665. [Google Scholar] [CrossRef]
- Zhu, H.; Chan, K.T.; Huang, X.; Cerra, C.; Blake, S.; Trigos, A.S.; Anderson, D.; Creek, D.J.; De Souza, D.P.; Wang, X.; et al. Cystathionine-beta-synthase is essential for AKT-induced senescence and suppresses the development of gastric cancers with PI3K/AKT activation. eLife 2022, 11, e71929. [Google Scholar] [CrossRef]
- Ni, S.J.; Yao, Z.Y.; Wei, X.; Heng, X.; Qu, S.Y.; Zhao, X.; Qi, Y.Y.; Ge, P.Y.; Xu, C.P.; Yang, N.Y.; et al. Vagus nerve stimulated by microbiota-derived hydrogen sulfide mediates the regulation of berberine on microglia in transient middle cerebral artery occlusion rats. Phytother. Res. 2022, 36, 2964–2981. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Condition/ Enzyme | Hypoxia | Hyperoxia | ROS | NO∙ | CO | H2S |
|---|---|---|---|---|---|---|
| Ceramides | ↑OL [52], CM [53], HC [54], PA [55] ↓VSMC [56] | ↑cancer cells [82,83,89], EC [90,91], MC [91], CM [53] | ↑MC [91,138], EC [91,139] ↓DC [140], U937 [141,142] | ↑cancer cells [165] | ||
| Sphingosine | ↑cortex [167] | |||||
| S1P | ↑VSMC [56], EC [59], cancer cells [58,61] | ↑mouse lung [77], human lung [78], EC [77] | ↓CM [121] | |||
| SM, Gangliosides | ↓brain [157,158] | |||||
| Cholesterolesters | ↑brain [157] | |||||
| Cerebrosides | ↓OL [52] | ↓brain [157] | ||||
| nSMase | ↑CM [53], PA [55] | ↑EC [90], CM [53] | ↑MC [138] | |||
| aSMase | ↑HC [54] | ↑EC [90] | ↑MC [138] ↓DC [140], U937 [141,142] | ↑cancer cells [165] | ||
| nCDase, aCDase | ↓MC [138,147] | |||||
| ACER2 | ↑adipocytes [57] | |||||
| Sphk1 | ↑EC [59], PSMC [60], cancer cells [58,62] | ↑mouse lung [77,79] | ↓CM [121] | |||
| Sphk2 | ↑cancer cells [61], PSMC [60] ↓EC [59] | |||||
| SPL | Tyr nitration [131] | |||||
| SPT2 | ↑neuroblastoma cells [69] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huwiler, A.; Beck, K.-F.; Pfeilschifter, J. Cross-Regulation of the Cellular Redox System, Oxygen, and Sphingolipid Signalling. Metabolites 2023, 13, 426. https://doi.org/10.3390/metabo13030426
Huwiler A, Beck K-F, Pfeilschifter J. Cross-Regulation of the Cellular Redox System, Oxygen, and Sphingolipid Signalling. Metabolites. 2023; 13(3):426. https://doi.org/10.3390/metabo13030426
Chicago/Turabian StyleHuwiler, Andrea, Karl-Friedrich Beck, and Josef Pfeilschifter. 2023. "Cross-Regulation of the Cellular Redox System, Oxygen, and Sphingolipid Signalling" Metabolites 13, no. 3: 426. https://doi.org/10.3390/metabo13030426
APA StyleHuwiler, A., Beck, K.-F., & Pfeilschifter, J. (2023). Cross-Regulation of the Cellular Redox System, Oxygen, and Sphingolipid Signalling. Metabolites, 13(3), 426. https://doi.org/10.3390/metabo13030426