


Comparative Transcriptome Analysis Reveals the Underlying Response Mechanism to Salt Stress in Maize Seedling Roots


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Treatment
2.2. Measurements of Physiological Parameters
2.3. Sample Collection and Acquisition of Transcriptome Data
2.4. RNA Extraction and RNA-Seq
2.5. Trend Analysis
2.6. GO and KEGG Enrichment Analysis
2.7. Quantitative Real-Time PCR
2.8. Statistical Analysis
3. Results
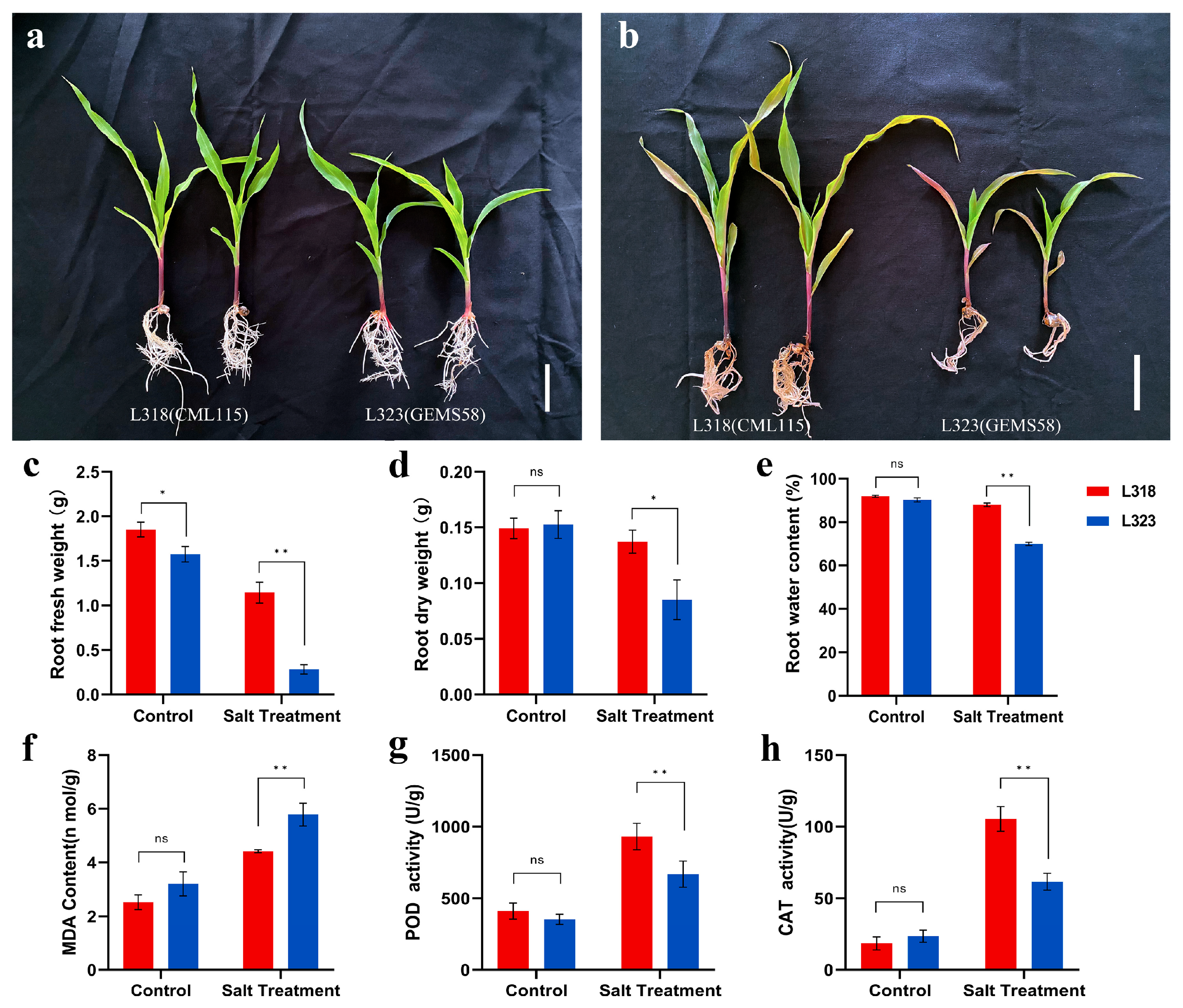
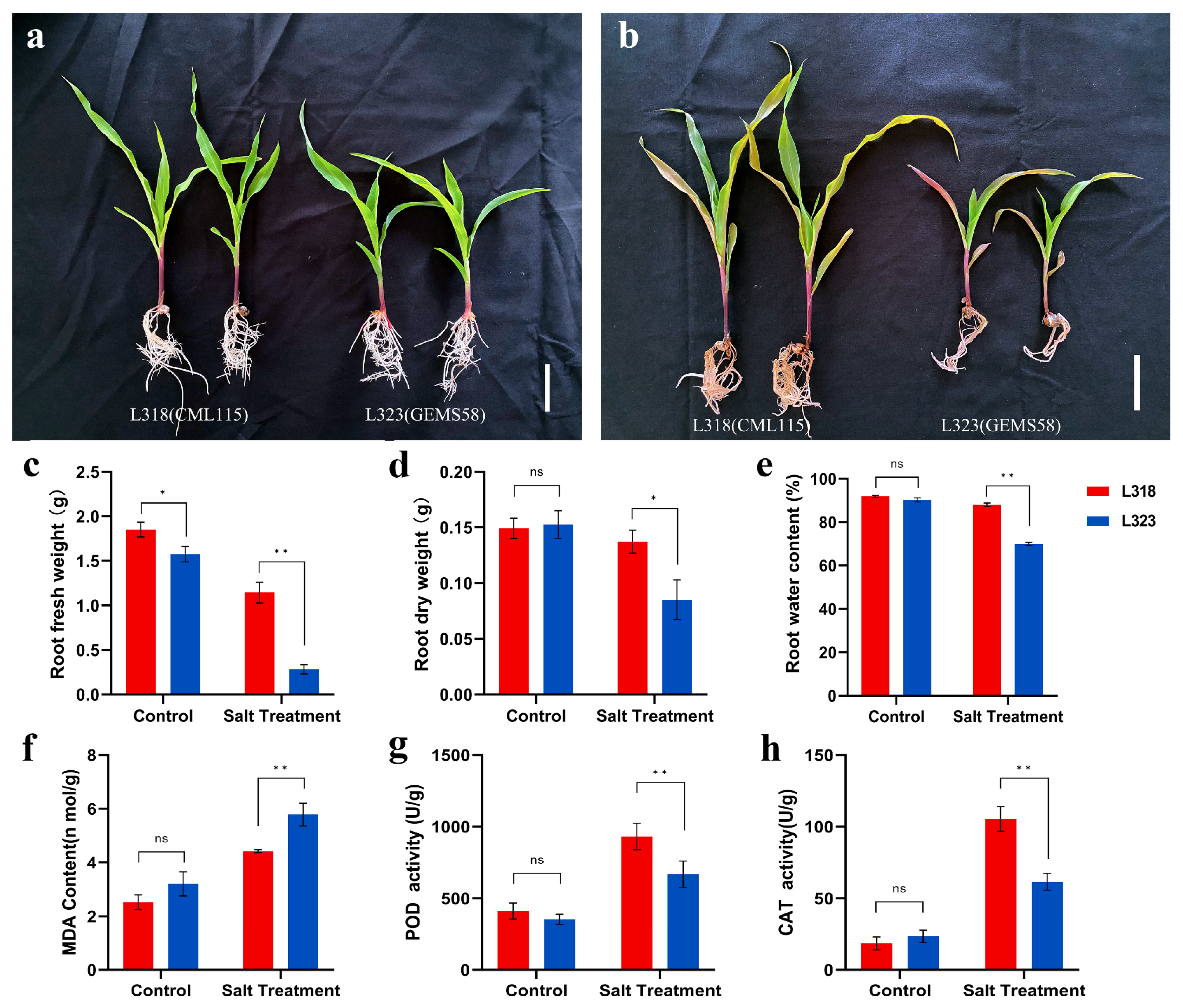
3.1. Phenotypic and Physiological Responses of Two Inbred Maize Lines to Salt Stress
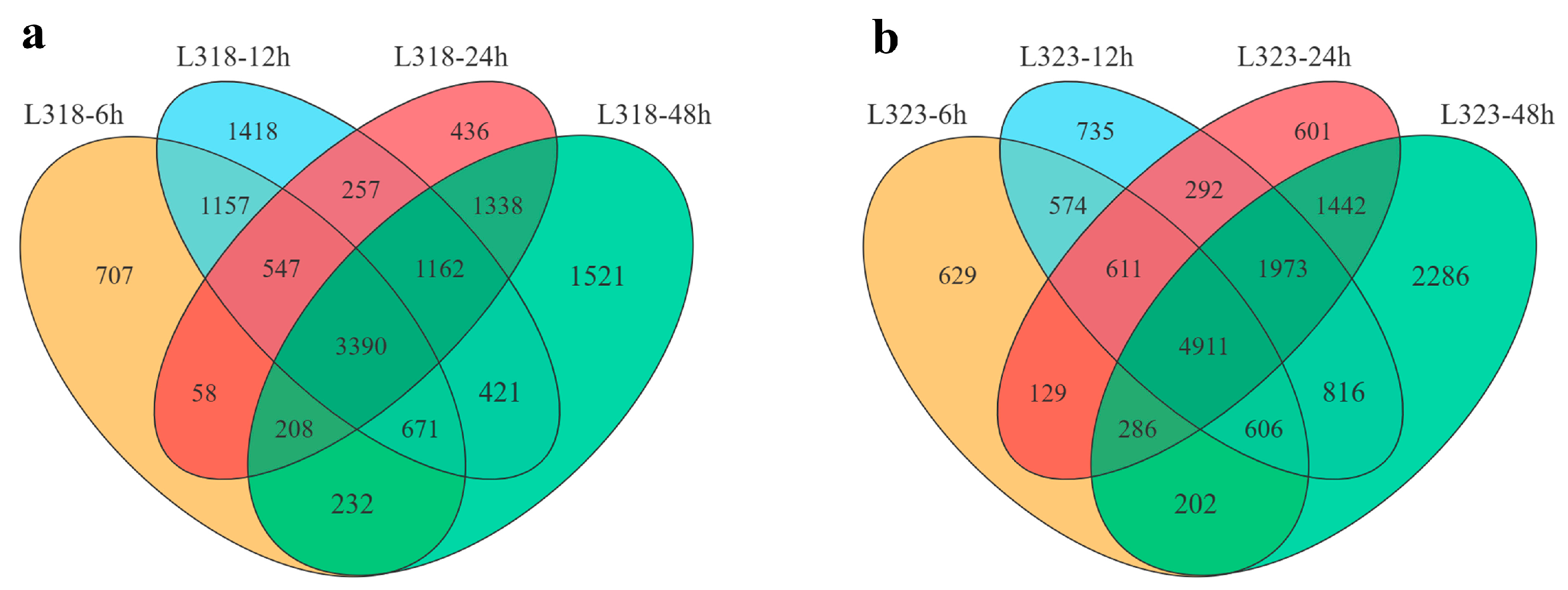
3.2. Transcriptome Analysis
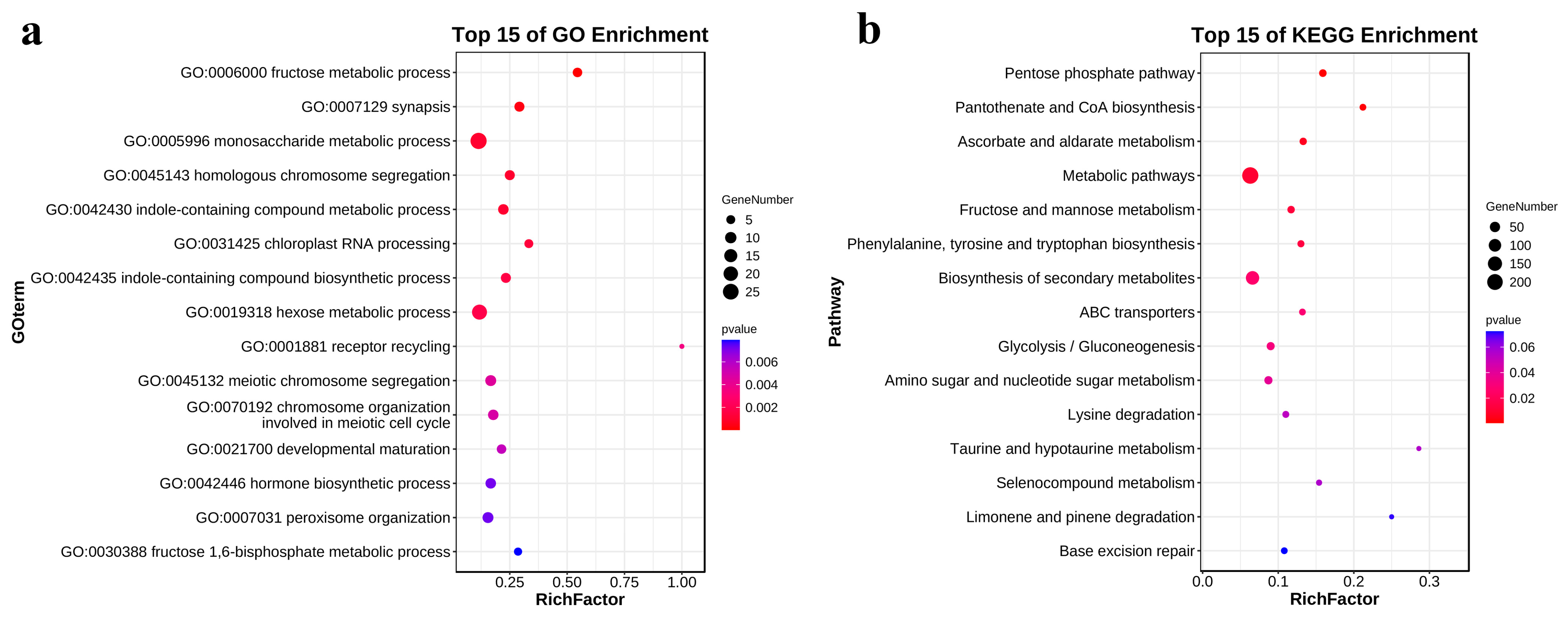
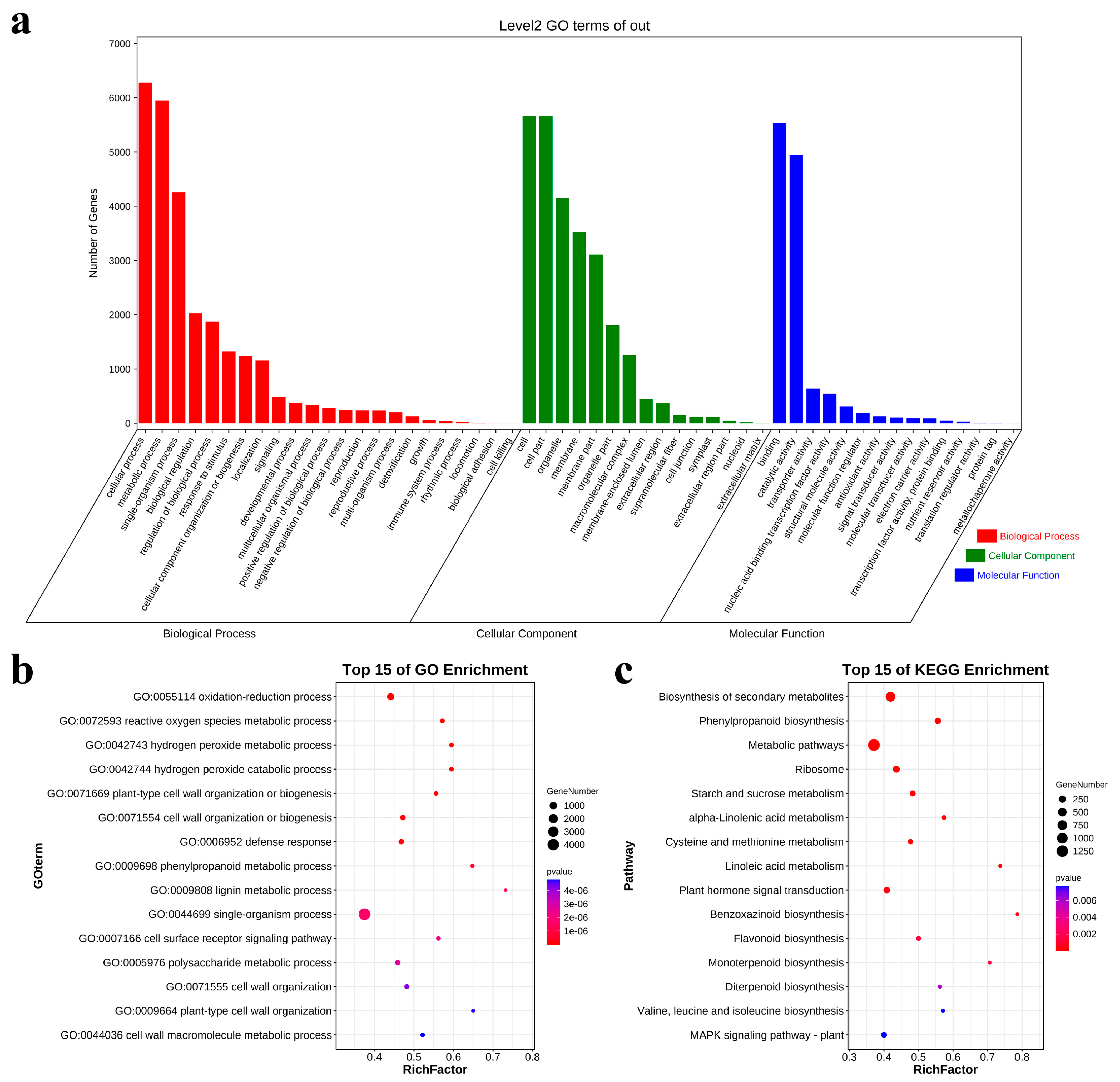
3.3. GO and KEGG Enrichment Analysis of DEGs Involved in Salt Tolerance Response
3.4. Analysis of the Expression Trend of Salt-Tolerant Genes
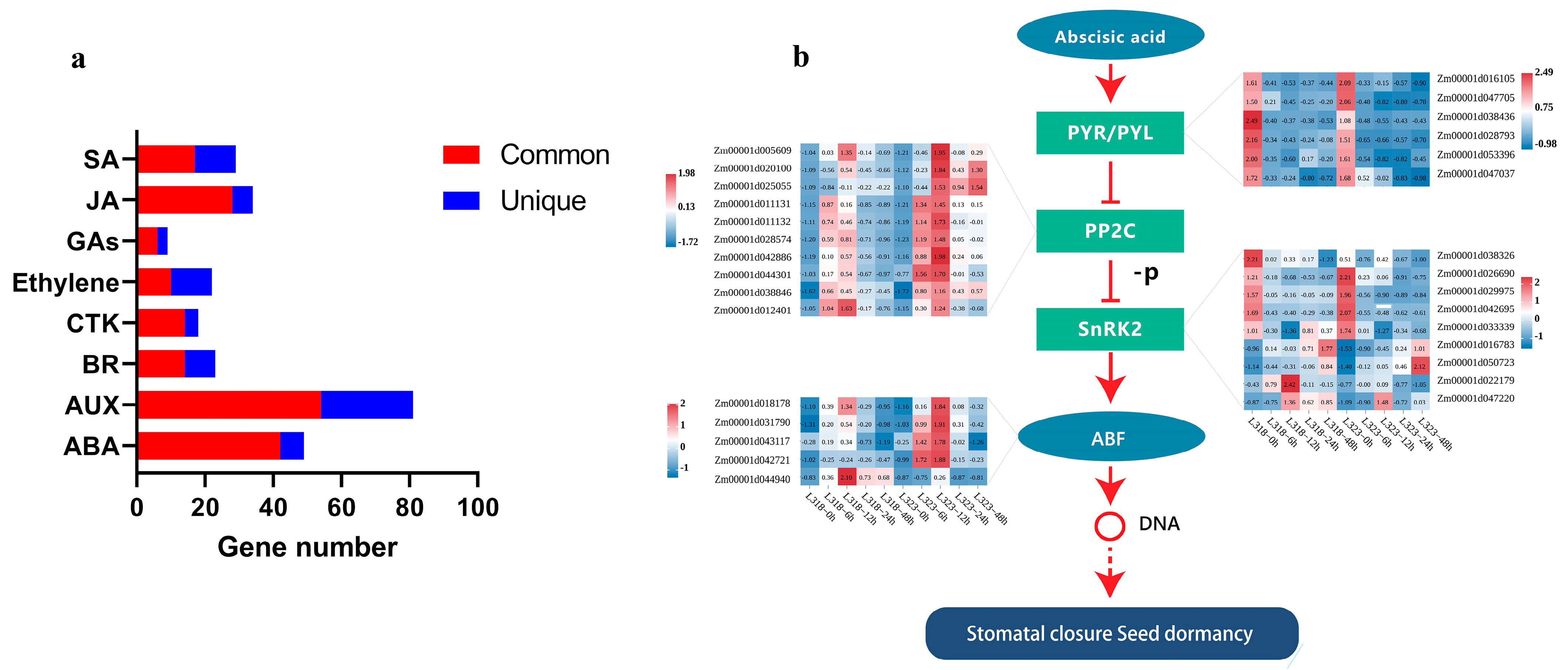
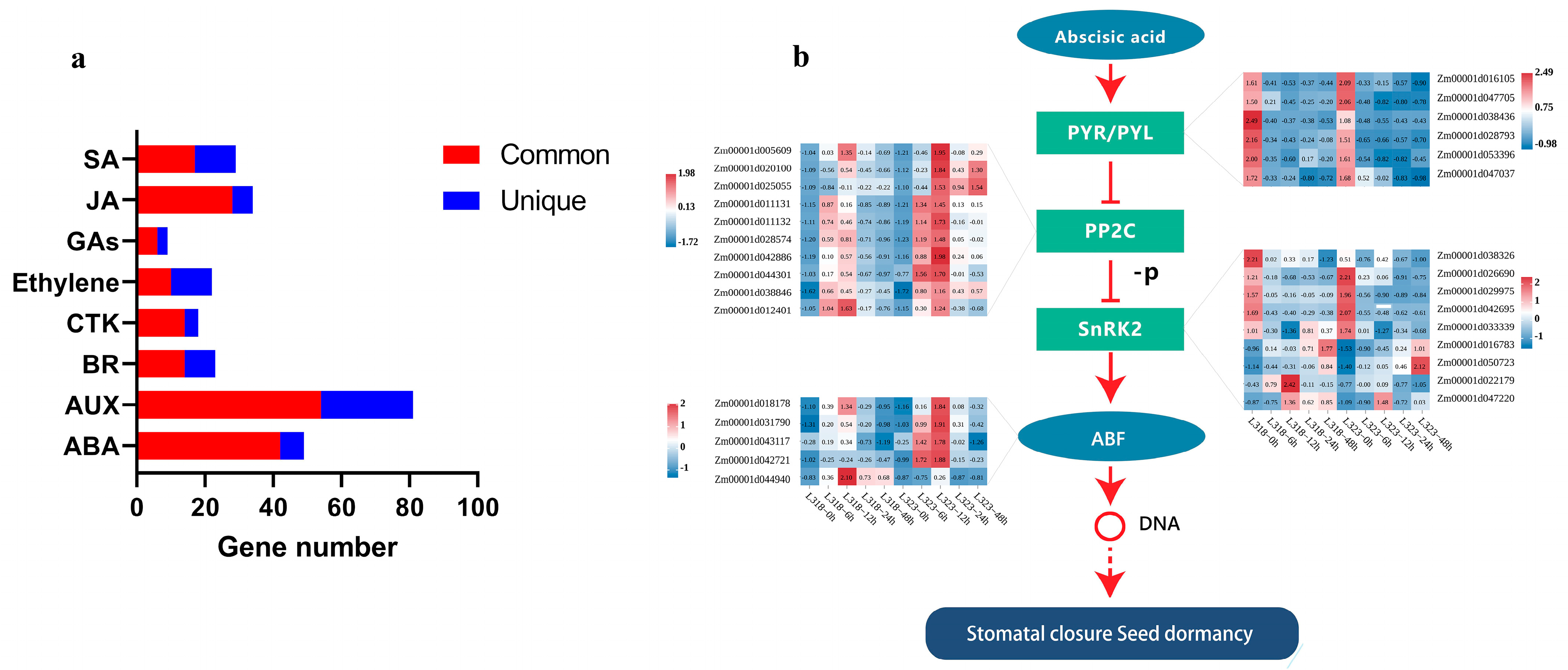
3.5. DEGs Involved in Plant Hormone Signal Transduction in Maize Root
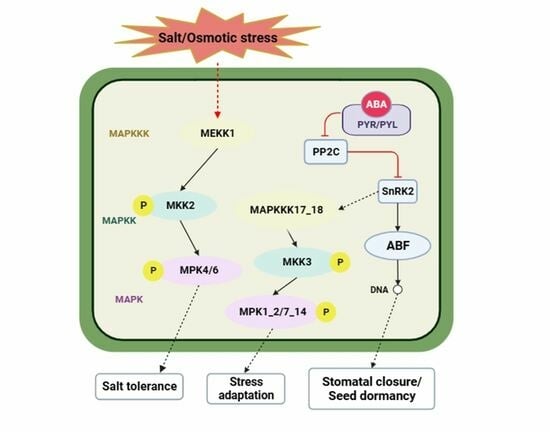
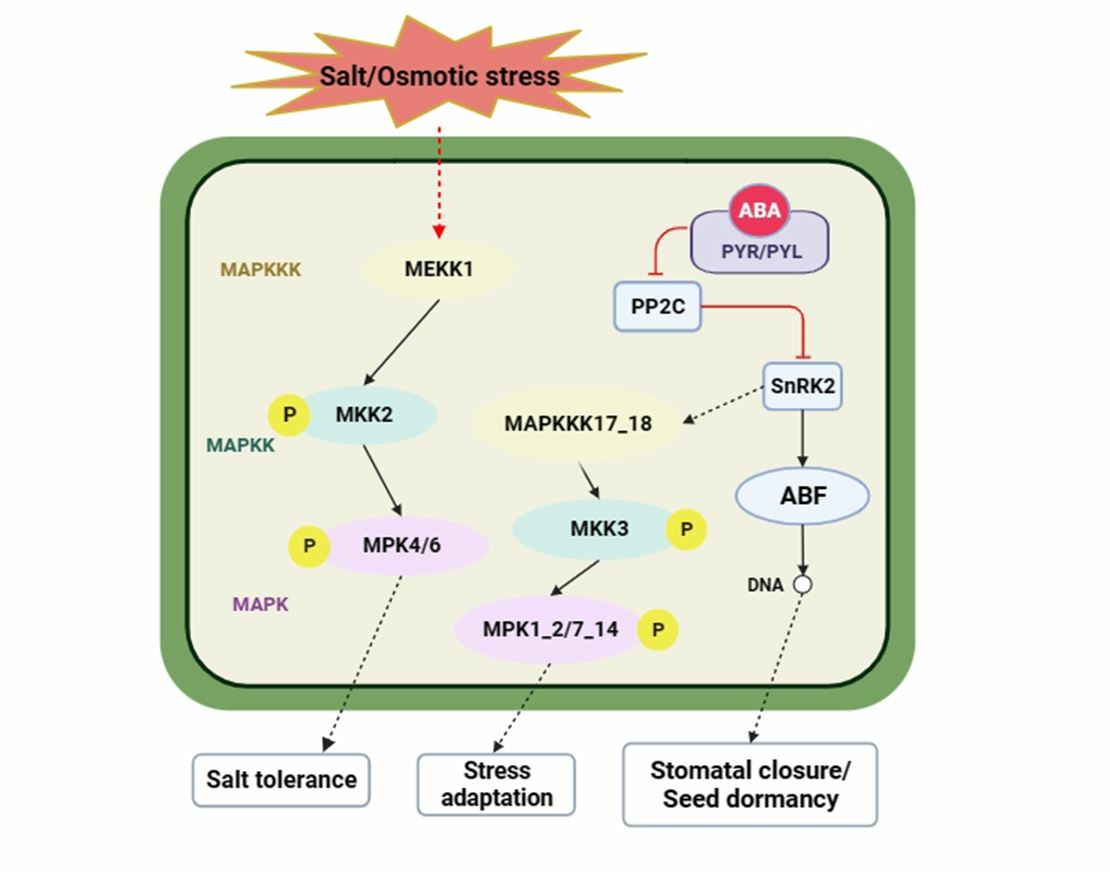
3.6. DEGs Involved in the MAPK Signaling Pathway–Plant in Maize Roots
3.7. Changes in Differentially Expressed TFs under Salt Stress
3.8. Validation of Candidate Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ismail, A.M.; Horie, T. Genomics, Physiology, and Molecular Breeding Approaches for Improving Salt Tolerance. Annu. Rev. Plant Biol. 2017, 68, 405–434. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhao, H.; Ren, L.; Song, W.; Zeng, B.; Guo, J.; Wang, B.; Liu, Z.; Chen, J.; Li, W.; et al. Genome-wide genetic changes during modern breeding of maize. Nat. Genet. 2012, 44, 812–815. [Google Scholar] [CrossRef]
- van Zelm, E.; Zhang, Y.; Testerink, C. Salt Tolerance Mechanisms of Plants. Annu. Rev. Plant Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef]
- Yu, Z.; Duan, X.; Luo, L.; Dai, S.; Ding, Z.; Xia, G. How Plant Hormones Mediate Salt Stress Responses. Trends Plant Sci. 2020, 25, 1117–1130. [Google Scholar] [CrossRef]
- Hake, S.; Ross-Ibarra, J. Genetic, evolutionary and plant breeding insights from the domestication of maize. Elife 2015, 4, e05861. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; James, R.A.; Xu, B.; Athman, A.; Conn, S.J.; Jordans, C.; Byrt, C.S.; Hare, R.A.; Tyerman, S.D.; Tester, M.; et al. Wheat grain yield on saline soils is improved by an ancestral Na⁺ transporter gene. Nat. Biotechnol. 2012, 30, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, D.; Pudussery, M.V.; Kumar, R.; Pallete, A.; Markley, P.; Bridges, W.C.; Sekhon, R.S. Characterization of natural genetic variation identifies multiple genes involved in salt tolerance in maize. Funct. Integr. Genom. 2020, 20, 261–275. [Google Scholar] [CrossRef]
- Deinlein, U.; Stephan, A.B.; Horie, T.; Luo, W.; Xu, G.; Schroeder, J.I. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014, 19, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Munns, R. Genes and salt tolerance: Bringing them together. New Phytol. 2005, 167, 645–663. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, P.; Abdel Latef, A.A.; Hashem, A.; Abd Allah, E.F.; Gucel, S.; Tran, L.S. Nitric Oxide Mitigates Salt Stress by Regulating Levels of Osmolytes and Antioxidant Enzymes in Chickpea. Front. Plant Sci. 2016, 7, 347. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, J.; Hu, J.; Xiong, W.; Du, C.; Lu, M. Integrated regulatory network reveals the early salt tolerance mechanism of Populus euphratica. Sci. Rep. 2017, 7, 6769. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Ma, X.; Wan, P.; Liu, L. Plant salt-tolerance mechanism: A review. Biochem. Biophys. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Liang, X.; Zhao, H.; Xu, Z.; Chen, L.; Yang, X.; Qin, F.; Zhang, J.; Jiang, C. Cytokinin signaling promotes salt tolerance by modulating shoot chloride exclusion in maize. Mol. Plant 2023, 16, 1031–1047. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhu, C.; Wang, C.; Liu, L.; Shen, Q.; Xu, D.; Wang, Q. Maize transcription factor ZmEREB20 enhanced salt tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2021, 159, 257–267. [Google Scholar] [CrossRef]
- Bo, C.; Chen, H.; Luo, G.; Li, W.; Zhang, X.; Ma, Q.; Cheng, B.; Cai, R. Maize WRKY114 gene negatively regulates salt-stress tolerance in transgenic rice. Plant Cell Rep. 2020, 39, 135–148. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, Y.; Zhao, X.; Wang, J.; Gu, M.; Yuan, Z. Transcriptomic Profiling of Pomegranate Provides Insights into Salt Tolerance. Agronomy 2020, 10, 44. [Google Scholar] [CrossRef]
- Cui, D.; Wu, D.; Somarathna, Y.; Xu, C.; Li, S.; Li, P.; Zhang, H.; Chen, H.; Zhao, L. QTL mapping for salt tolerance based on snp markers at the seedling stage in maize (Zea mays L.). Euphytica 2015, 203, 273–283. [Google Scholar] [CrossRef]
- Zhang, M.; Cao, Y.; Wang, Z.; Wang, Z.Q.; Shi, J.; Liang, X.; Song, W.; Chen, Q.; Lai, J.; Jiang, C. A retrotransposon in an HKT1 family sodium transporter causes variation of leaf Na(+) exclusion and salt tolerance in maize. New Phytol. 2018, 217, 1161–1176. [Google Scholar] [CrossRef]
- Sharmin, R.A.; Bhuiyan, M.R.; Lv, W.; Yu, Z.; Chang, F.; Kong, J.; Bhat, J.A.; Zhao, T. RNA-Seq based transcriptomic analysis revealed genes associated with seed-flooding tolerance in wild soybean (Glycine soja Sieb. & Zucc.). Environ. Exp. Bot. 2020, 171, 103906. [Google Scholar] [CrossRef]
- Wang, H.; Miyazaki, S.; Kawai, K.; Deyholos, M.; Galbraith, D.W.; Bohnert, H.J. Temporal progression of gene expression responses to salt shock in maize roots. Plant Mol. Biol. 2003, 52, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shang, X.; Sun, M.; Tang, S.; Khan, A.; Zhang, D.; Yan, H.; Jiang, Y.; Yu, F.; Wu, Y.; et al. Comparative Transcriptome Analysis of Two Sweet Sorghum Genotypes with Different Salt Tolerance Abilities to Reveal the Mechanism of Salt Tolerance. Int. J. Mol. Sci. 2022, 23, 2272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, J.; Huang, Y.; Wu, H.; Hu, X.; Cheng, B.; Ma, Q.; Zhao, Y. Comparative Transcriptomics Reveals the Molecular Mechanism of the Parental Lines of Maize Hybrid An’nong876 in Response to Salt Stress. Int. J. Mol. Sci. 2022, 23, 5231. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Razzaq, A.; Ali, A.; Safdar, L.B.; Zafar, M.M.; Rui, Y.; Shakeel, A.; Shaukat, A.; Ashraf, M.; Gong, W.; Yuan, Y. Salt stress induces physiochemical alterations in rice grain composition and quality. J. Food Sci. 2020, 85, 14–20. [Google Scholar] [CrossRef]
- Hakim, M.A.; Juraimi, A.S.; Hanafi, M.M.; Ismail, M.R.; Selamat, A.; Rafii, M.Y.; Latif, M.A. Biochemical and anatomical changes and yield reduction in rice (Oryza sativa L.) under varied salinity regimes. Biomed. Res. Int. 2014, 2014, 208584. [Google Scholar] [CrossRef]
- Saini, S.; Sharma, I.; Kaur, N.; Pati, P.K. Auxin: A master regulator in plant root development. Plant Cell Rep. 2013, 32, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Jagendorf, A.; Zhu, J.-K. Understanding and Improving Salt Tolerance in Plants. Crop Sci. 2005, 45, 437–448. [Google Scholar] [CrossRef]
- Fan, X.; Jiang, H.; Meng, L.; Chen, J. Gene Mapping, Cloning and Association Analysis for Salt Tolerance in Rice. Int. J. Mol. Sci. 2021, 22, 11674. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, C.D.; Lim, S.; Salzman, R.A.; Kagiampakis, I.; Morishige, D.T.; Weers, B.D.; Klein, R.R.; Pratt, L.H.; Cordonnier-Pratt, M.M.; Klein, P.E.; et al. Sorghum bicolor’s transcriptome response to dehydration, high salinity and ABA. Plant Mol. Biol. 2005, 58, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Sui, N.; Yang, Z.; Liu, M.; Wang, B. Identification and transcriptomic profiling of genes involved in increasing sugar content during salt stress in sweet sorghum leaves. BMC Genom. 2015, 16, 534. [Google Scholar] [CrossRef] [PubMed]
- Ukwatta, J.; Pabuayon, I.C.M.; Park, J.; Chen, J.; Chai, X.; Zhang, H.; Zhu, J.K.; Xin, Z.; Shi, H. Comparative physiological and transcriptomic analysis reveals salinity tolerance mechanisms in Sorghum bicolor (L.) Moench. Planta 2021, 254, 98. [Google Scholar] [CrossRef] [PubMed]
- Seif El-Yazal, S.A.; Seif El-Yazal, M.A.; Dwidar, E.F.; Rady, M.M. Phytohormone crosstalk research: Cytokinin and its crosstalk with other phytohormones. Curr. Protein Pept. Sci. 2015, 16, 395–405. [Google Scholar] [CrossRef]
- Finkelstein, R. Abscisic Acid synthesis and response. Arab. Book 2013, 11, e0166. [Google Scholar] [CrossRef]
- Belda-Palazón, B.; Adamo, M.; Valerio, C.; Ferreira, L.J.; Confraria, A.; Reis-Barata, D.; Rodrigues, A.; Meyer, C.; Rodriguez, P.L.; Baena-González, E. A dual function of SnRK2 kinases in the regulation of SnRK1 and plant growth. Nat. Plants 2020, 6, 1345–1353. [Google Scholar] [CrossRef]
- Lv, B.; Yan, Z.; Tian, H.; Zhang, X.; Ding, Z. Local Auxin Biosynthesis Mediates Plant Growth and Development. Trends Plant Sci. 2019, 24, 6–9. [Google Scholar] [CrossRef]
- Liu, C.; Mao, B.; Yuan, D.; Chu, C.; Duan, M. Salt tolerance in rice: Physiological responses and molecular mechanisms. Crop J. 2022, 10, 13–25. [Google Scholar] [CrossRef]
- Amirbakhtiar, N.; Ismaili, A.; Ghaffari, M.R.; Mirdar Mansuri, R.; Sanjari, S.; Shobbar, Z.S. Transcriptome analysis of bread wheat leaves in response to salt stress. PLoS ONE 2021, 16, e0254189. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhao, H.; Sun, L.; Wu, W.; Li, C.; Wu, Q. Genome-wide identification of MAPK gene family members in Fagopyrum tataricum and their expression during development and stress responses. BMC Genom. 2022, 23, 96. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Feng, L.; Liu, Y.; Liao, W. Mitogen-Activated Protein Kinase Is Involved in Salt Stress Response in Tomato (Solanum lycopersicum) Seedlings. Int. J. Mol. Sci. 2022, 23, 7645. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Z.; Chen, C.; Xu, M. The MKK2a Gene Involved in the MAPK Signaling Cascades Enhances Populus Salt Tolerance. Int. J. Mol. Sci. 2022, 23, 10185. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, M.; Gao, Z.; Ren, W.; Yang, F.; He, H.; Zhao, J. RNA-Seq Analysis Reveals MAPKKK Family Members Related to Drought Tolerance in Maize. PLoS ONE 2015, 10, e0143128. [Google Scholar] [CrossRef]
- Cheng, Z.; Luan, Y.; Meng, J.; Sun, J.; Tao, J.; Zhao, D. WRKY Transcription Factor Response to High-Temperature Stress. Plants 2021, 10, 2211. [Google Scholar] [CrossRef]
- Zhang, B.; Feng, C.; Chen, L.; Li, B.; Zhang, X.; Yang, X. Identification and Functional Analysis of bZIP Genes in Cotton Response to Drought Stress. Int. J. Mol. Sci. 2022, 23, 14894. [Google Scholar] [CrossRef]
- Jin, R.; Kim, H.S.; Yu, T.; Zhang, A.; Yang, Y.; Liu, M.; Yu, W.; Zhao, P.; Zhang, Q.; Cao, Q.; et al. Identification and function analysis of bHLH genes in response to cold stress in sweetpotato. Plant Physiol. Biochem. 2021, 169, 224–235. [Google Scholar] [CrossRef]
- Waititu, J.K.; Zhang, X.; Chen, T.; Zhang, C.; Zhao, Y.; Wang, H. Transcriptome Analysis of Tolerant and Susceptible Maize Genotypes Reveals Novel Insights about the Molecular Mechanisms Underlying Drought Responses in Leaves. Int. J. Mol. Sci. 2021, 22, 6980. [Google Scholar] [CrossRef]
- Mao, H.; Yu, L.; Han, R.; Li, Z.; Liu, H. ZmNAC55, a maize stress-responsive NAC transcription factor, confers drought resistance in transgenic Arabidopsis. Plant Physiol. Biochem. 2016, 105, 55–66. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Chen, B.; Zhang, P.; Han, Q.; Zhao, G.; Zhao, F. Comparative Transcriptome Analysis Reveals the Underlying Response Mechanism to Salt Stress in Maize Seedling Roots. Metabolites 2023, 13, 1155. https://doi.org/10.3390/metabo13111155
Zhang C, Chen B, Zhang P, Han Q, Zhao G, Zhao F. Comparative Transcriptome Analysis Reveals the Underlying Response Mechanism to Salt Stress in Maize Seedling Roots. Metabolites. 2023; 13(11):1155. https://doi.org/10.3390/metabo13111155
Chicago/Turabian StyleZhang, Chen, Bin Chen, Ping Zhang, Qinghui Han, Guangwu Zhao, and Fucheng Zhao. 2023. "Comparative Transcriptome Analysis Reveals the Underlying Response Mechanism to Salt Stress in Maize Seedling Roots" Metabolites 13, no. 11: 1155. https://doi.org/10.3390/metabo13111155
APA StyleZhang, C., Chen, B., Zhang, P., Han, Q., Zhao, G., & Zhao, F. (2023). Comparative Transcriptome Analysis Reveals the Underlying Response Mechanism to Salt Stress in Maize Seedling Roots. Metabolites, 13(11), 1155. https://doi.org/10.3390/metabo13111155