
Genome-Wide Transcriptome Analysis Revealing the Genes Related to Sugar Metabolism in Kernels of Sweet Corn

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Field Planting and Sampling of Sweet Corn
2.2. Determination of WSP in Developing Kernels
2.3. RNA Extraction and RNA-Seq
2.4. DEGs Identification
2.5. Time Course Analysis
2.6. Weighted Correlation Network Analysis
2.7. RT-qPCR
2.8. Statistical Analysis
3. Results
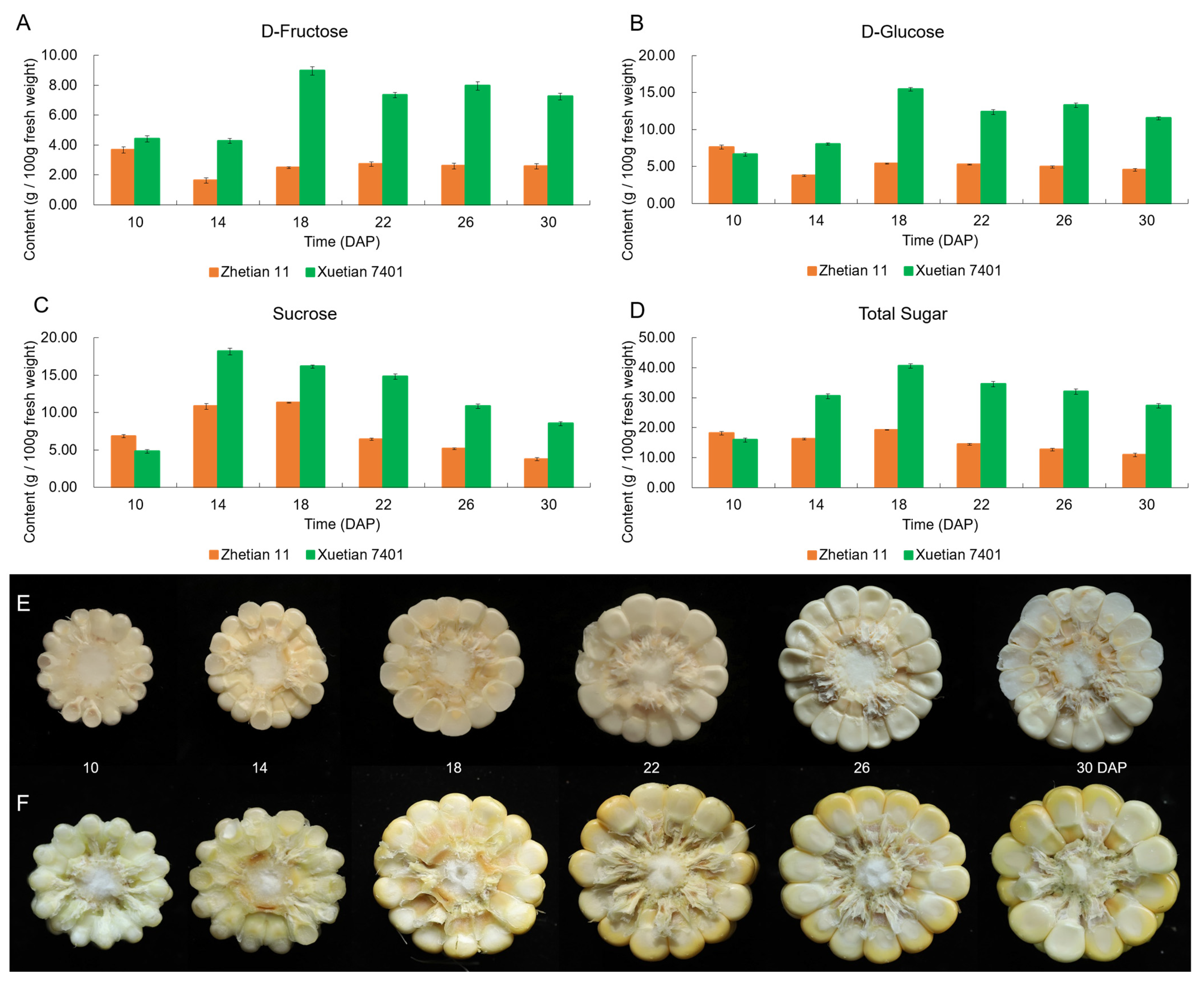
3.1. Dynamic Changes of WSP Contents in Developing Kernels
3.2. Quality Analysis of RNA-Seq Data
3.3. RNA-Seq Analysis and DEG Statistics
3.4. RNA-Seq Analysis and DEG Statistics
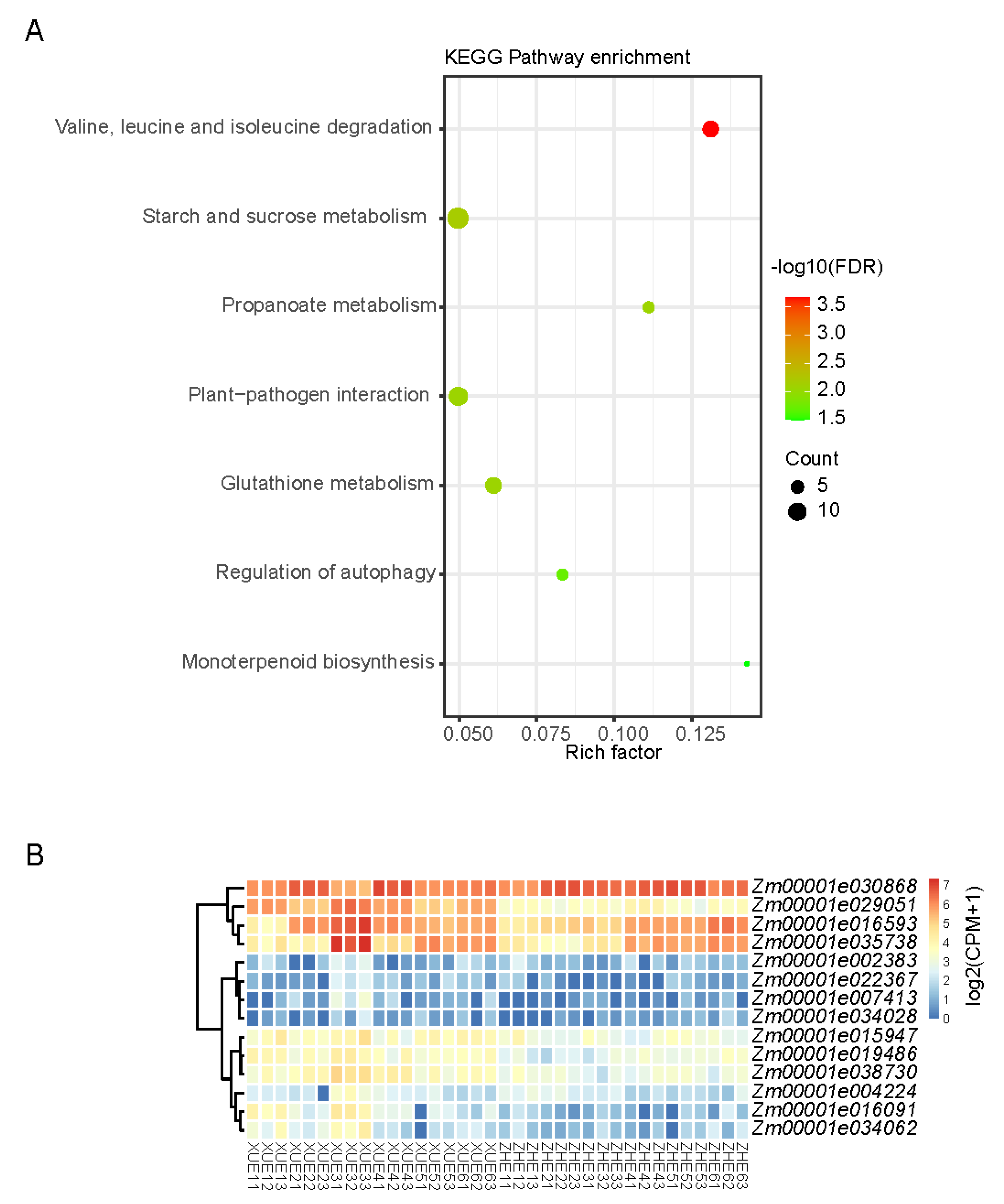
3.5. Identification of Genes Related to Sugar Accumulation via Weighted Correlation Network Analysis (WGCNA)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mir, H.R.; Yadav, S.K.; Yadav, S. Hydropriming associated physiological and biochemical changes responsible for the enhanced planting value of maize hybrid and its parental line seeds. Turk. J. Agric. For. 2021, 45, 335–348. [Google Scholar] [CrossRef]
- Nawaz, H.; Hussain, N.; Jamil, M.; Yasmeen, A.; Bukhari, S.A.H.; Aurangzaib, M.; Usman, M. Seed biopriming mitigates terminal drought stress at reproductive stage of maize by enhancing gas exchange attributes and nutrient uptake. Turk. J. Agric. For. 2020, 44, 250–261. [Google Scholar] [CrossRef]
- FAO (Food and Agriculture Organization of the United Nations). FAOSTAT: FAO Statistical Databases; FAO: Rome, Italy, 2021. [Google Scholar]
- Popalia, C.; Kumar, N. Effect of Temperature and Processing Time on Physico-Chemical Characteristics in Hot Water Blanching of Sweet Corn Kernels. J. Inst. Eng. (India) Ser. A 2021, 102, 163–173. [Google Scholar] [CrossRef]
- USDA (U.S. Department of Agriculture). National Agricultural Statistics Service: Quick Stats Tools; USDA: Washington, DC, USA, 2021.
- Rahman, A.; Wong, K.; Jane, J.; Myers, A.M.; James, M.G. Characterization of SU1 isoamylase, a determinant of storage starch structure in maize. Plant Physiol. 1998, 117, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Creech, R.G. Genetic Control of Carbohydrate Synthesis in Maize Endosperm. Genetics 1965, 52, 1175–1186. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Nelson, O.E. Starch-deficient maize mutant lacking adenosine dephosphate glucose pyrophosphorylase activity. Science 1966, 151, 341–343. [Google Scholar] [CrossRef]
- Tracy, W.F. History, Genetics, and Breeding of Supersweet (shrunken2) Sweet Corn. Plant Breed. Rev. 1996, 14, 189–236. [Google Scholar]
- Boyer, C.; Shannon, J. The use of endosperm genes for sweet corn improvement. Plant Breed. Rev. 1984, 1, 161–193. [Google Scholar]
- Singh, I.; Langyan, S.; Yadava, P. Sweet Corn and Corn-Based Sweeteners. Sugar Tech 2014, 16, 144–149. [Google Scholar] [CrossRef]
- Ferguson, J.E.; Rhodes, A.M.; Dickinson, D.B. The genetics of sugary enhancer (se), an independent modifier of sweet corn (su). J. Hered. 1978, 69, 377–380. [Google Scholar] [CrossRef]
- Laughnan, J. The Effect of the sh2 Factor on Carbohydrate Reserves in the Mature Endosperm of Maize. Genetics 1953, 38, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Denyer, K.; Martin, C. What Controls the Amount and Structure of Starch in Storage Organs? Plant Physiol. 1995, 107, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Szymanek, M.; Tanaś, W.; Kassar, F. Kernel Carbohydrates Concentration in Sugary-1, Sugary Enhanced and Shrunken Sweet Corn Kernels. Agric. Agric. Sci. Procedia 2015, 7, 260–264. [Google Scholar] [CrossRef]
- Finegan, C.; Boehlein, S.K.; Leach, K.A.; Madrid, G.; Hannah, L.C.; Koch, K.E.; Tracy, W.F.; Resende, M.F.R., Jr. Genetic Perturbation of the Starch Biosynthesis in Maize Endosperm Reveals Sugar-Responsive Gene Networks. Front. Plant Sci. 2021, 12, 800326. [Google Scholar] [CrossRef] [PubMed]
- Smidansky, E.D.; Clancy, M.; Meyer, F.D.; Lanning, S.P.; Blake, N.K.; Talbert, L.E.; Giroux, M.J. Enhanced ADP-glucose pyrophosphorylase activity in wheat endosperm increases seed yield. Proc. Natl. Acad. Sci. USA 2002, 99, 1724–1729. [Google Scholar] [CrossRef]
- Muir, J.G.; Rose, R.; Rosella, O.; Liels, K.; Barrett, J.S.; Shepherd, S.J.; Gibson, P.R. Measurement of short-chain carbohydrates in common Australian vegetables and fruits by high-performance liquid chromatography (HPLC). J. Agric. Food Chem. 2009, 57, 554–565. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wann, E. Leaching of Metabolites during Imbibition of Sweet Corn Seed of Different Endosperm Genotypes. Crop Sci. 1986, 26, 731–733. [Google Scholar] [CrossRef]
- Chourey, P.S.; Nelson, O.E. The enzymatic deficiency conditioned by the shrunken-1 mutations in maize. Biochem. Genet. 1976, 14, 1041–1055. [Google Scholar] [CrossRef]
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic O2 evolution-I. A linear four step mechanism. Photochem. Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef]
- Loll, B.; Kern, J.; Saenger, W.; Zouni, A.; Biesiadka, J. Towards complete cofactor arrangement in the 3.0 A resolution structure of photosystem II. Nature 2005, 438, 1040–1044. [Google Scholar] [CrossRef]
- Ke, B. Photosynthesis: Photobiochemistry and Photobiophysics; Kluwer Academic Publishers: Urbana, IL, USA, 2001. [Google Scholar]
- Berthold, D.A.; Babcock, G.T.; Yocum, C.F. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 1981, 134, 231–234. [Google Scholar] [CrossRef]
- Kim, S.; Sandusky, P.; Bowlby, N.R.; Aebersold, R.; Green, B.R.; Vlahakis, S.; Yocum, C.F.; Pichersky, E. Characterization of a spinach psbS cDNA encoding the 22 kDa protein of photosystem II. FEBS Lett. 1992, 314, 67–71. [Google Scholar] [CrossRef]
- Wedel, N.; Klein, R.; Ljungberg, U.; Andersson, B.; Herrmann, R.G. The single-copy gene psbS codes for a phylogenetically intriguing 22 kDa polypeptide of photosystem II. FEBS Lett. 1992, 314, 61–66. [Google Scholar] [CrossRef]
- Kim, S.; Pichersky, E.; Yocum, C.F. Topological studies of spinach 22 kDa protein of Photosystem II. Biochim. Biophys. Acta 1994, 1188, 339–348. [Google Scholar] [CrossRef]
- Li, X.P.; Bjorkman, O.; Shih, C.; Grossman, A.R.; Rosenquist, M.; Jansson, S.; Niyogi, K.K. A pigment-binding protein essential for regulation of photosynthetic light harvesting. Nature 2000, 403, 391–395. [Google Scholar] [CrossRef]
- Kulheim, C.; Agren, J.; Jansson, S. Rapid regulation of light harvesting and plant fitness in the field. Science 2002, 297, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Kromdijk, J.; Glowacka, K.; Leonelli, L.; Gabilly, S.T.; Iwai, M.; Niyogi, K.K.; Long, S.P. Improving photosynthesis and crop productivity by accelerating recovery from photoprotection. Science 2016, 354, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.L.; Dickinson, D.B. Hexokinase from maize endosperm and scutellum. Plant Physiol. 1973, 51, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Claeyssen, E.; Rivoal, J. Isozymes of plant hexokinase: Occurrence, properties and functions. Phytochemistry 2007, 68, 709–731. [Google Scholar] [CrossRef] [PubMed]
- Pego, J.V.; Weisbeek, P.J.; Smeekens, S.C. Mannose inhibits Arabidopsis germination via a hexokinase-mediated step. Plant Physiol. 1999, 119, 1017–1023. [Google Scholar] [CrossRef]
- Kim, H.B.; Cho, J.I.; Ryoo, N.; Shin, D.H.; Park, Y.I.; Hwang, Y.S.; Lee, S.K.; An, G.; Jeon, J.S. Role of rice cytosolic hexokinase OsHXK7 in sugar signaling and metabolism. J. Integr. Plant Biol. 2016, 58, 127–135. [Google Scholar] [CrossRef]
- Kang, Y.; Outlaw, W.H., Jr.; Andersen, P.C.; Fiore, G.B. Guard-cell apoplastic sucrose concentration—A link between leaf photosynthesis and stomatal aperture size in the apoplastic phloem loader Vicia faba L. Plant Cell Environ. 2007, 30, 551–558. [Google Scholar] [CrossRef]
- Medeiros, D.B.; Perez Souza, L.; Antunes, W.C.; Araujo, W.L.; Daloso, D.M.; Fernie, A.R. Sucrose breakdown within guard cells provides substrates for glycolysis and glutamine biosynthesis during light-induced stomatal opening. Plant J. 2018, 94, 583–594. [Google Scholar] [CrossRef]
- Karthikeyan, A.S.; Varadarajan, D.K.; Jain, A.; Held, M.A.; Carpita, N.C.; Raghothama, K.G. Phosphate starvation responses are mediated by sugar signaling in Arabidopsis. Planta 2007, 225, 907–918. [Google Scholar] [CrossRef]
- Sarowar, S.; Lee, J.Y.; Ahn, E.R.; Pai, H.S. A role of hexokinases in plant resistance to oxidative stress and pathogen infection. J. Plant Biol. 2008, 51, 341–346. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Chen, Y.; Li, R.; Wang, H.; Ding, L.; Wei, J. Isolation, structural analysis, and expression characteristics of the maize (Zea mays L.) hexokinase gene family. Mol. Biol. Rep. 2014, 41, 6157–6166. [Google Scholar] [CrossRef]
- Menu, T.; Saglio, P.; Granot, D.; Dai, N.; Raymond, P.; Ricard, B. High hexokinase activity in tomato fruit perturbs carbon and energy metabolism and reduces fruit and seed size. Plant Cell Environ. 2004, 27, 89–98. [Google Scholar] [CrossRef]
- Xie, M.; Chen, J.; Qin, Q.; Jiang, G.; Sun, C.; Zhang, H.; Xu, H. Invertase and hexokinase regulate sugar accumulation in strawberry aggregates. J. Plant Physiol. Mol. Biol. 2007, 33, 213–218. [Google Scholar]
- Zhang, S.; Zhao, B.; Huang, X.; Qi, K. Pear hexokinase gene PbHXK1 and its application. 201410439245, 3 December 2014. [Google Scholar]
- Opassiri, R.; Pomthong, B.; Onkoksoong, T.; Akiyama, T.; Esen, A.; Ketudat Cairns, J.R. Analysis of rice glycosyl hydrolase family 1 and expression of Os4bglu12 beta-glucosidase. BMC Plant Biol. 2006, 6, 33. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequences (5′-3′) |
|---|---|
| CyanateF | GCTGGTGAGGAGGAGAAACA |
| CyanateR | CAGCAATCATGCCAGGTAGA |
| ONT.1.1-F | GAGTCCTCTGGAAGCTCTGTG |
| ONT.1.1-R | GGAGCAGCATCAAACAACCTG |
| ONT.10003.1-F | ATCCAATGCTGGGGGTGATG |
| ONT.10003.1-R | GATGGAGAGGGTATGCGAGC |
| ONT.10030.1-F | ACTTGGTTTGCCACCTTTGC |
| ONT.10030.1-R | GAATTGCTGCGGATTGGACC |
| ONT.10032.2-F | TTCCAGGAGCACTTCGATGC |
| ONT.10032.2-R | AGTCTAGTCGAGGCCGAAGT |
| ONT.10200.1-F | AGCGTATCCGTCTGAGCAAC |
| ONT.10200.1-R | GCCTCCAATGCACGTTCAAG |
| ONT.11355.1-F | GTTGTCGTTCTGTTCACCGC |
| ONT.11355.1-R | TTCAGGTTCGTTCACCAGCA |
| ONT.11394.1-F | CTCGGATCGTCGTCTCATGG |
| ONT.11394.1-R | AAGTCCACCACCTTGGGTTC |
| DEG Set | DEG Number | Upregulated | Downregulated |
|---|---|---|---|
| Xuetian 7401 (10 DAP) vs. Xuetian 7401 (14 DAP) | 2636 | 1352 | 1284 |
| Xuetian 7401 (10 DAP) vs. Xuetian 7401 (18 DAP) | 3693 | 2209 | 1484 |
| Xuetian 7401 (10 DAP) vs. Xuetian 7401 (22 DAP) | 3057 | 1545 | 1512 |
| Xuetian 7401 (10 DAP) vs. Xuetian 7401 (26 DAP) | 4169 | 2069 | 2100 |
| Xuetian 7401 (10 DAP) vs. Xuetian 7401 (30 DAP) | 4596 | 2374 | 2222 |
| Zhetian 11 (10 DAP) vs. Zhetian 11 (14 DAP) | 1186 | 576 | 610 |
| Zhetian 11 (10 DAP) vs. Zhetian 11 (18 DAP) | 2207 | 1409 | 798 |
| Zhetian 11 (10 DAP) vs. Zhetian 11 (22 DAP) | 1760 | 986 | 774 |
| Zhetian 11 (10 DAP) vs. Zhetian 11 (26 DAP) | 2265 | 1177 | 1088 |
| Zhetian 11 (10 DAP) vs. Zhetian 11 (30 DAP) | 2576 | 1530 | 1046 |
| Zhetian 11 (10 DAP) vs. Xuetian 7401 (10 DAP) | 3552 | 1630 | 1922 |
| Zhetian 11 (14 DAP) vs. Xuetian 7401 (14 DAP) | 2519 | 1282 | 1237 |
| Zhetian 11 (18 DAP) vs. Xuetian 7401 (18 DAP) | 4036 | 2189 | 1847 |
| Zhetian 11 (22 DAP) vs. Xuetian 7401 (22 DAP) | 2147 | 999 | 1148 |
| Zhetian 11 (26 DAP) vs. Xuetian 7401 (26 DAP) | 2862 | 1457 | 1405 |
| Zhetian 11 (30 DAP) vs. Xuetian 7401 (30 DAP) | 2487 | 1144 | 1343 |
| Total | 45,748 | 23,928 | 21,820 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.; Feng, S.; Hou, J.; Zhu, Y.; Bao, F.; Han, H.; Tan, H.; Wang, G.; Zhao, F. Genome-Wide Transcriptome Analysis Revealing the Genes Related to Sugar Metabolism in Kernels of Sweet Corn. Metabolites 2022, 12, 1254. https://doi.org/10.3390/metabo12121254
Chen B, Feng S, Hou J, Zhu Y, Bao F, Han H, Tan H, Wang G, Zhao F. Genome-Wide Transcriptome Analysis Revealing the Genes Related to Sugar Metabolism in Kernels of Sweet Corn. Metabolites. 2022; 12(12):1254. https://doi.org/10.3390/metabo12121254
Chicago/Turabian StyleChen, Bin, Shouli Feng, Junfeng Hou, Ying Zhu, Fei Bao, Hailiang Han, Heping Tan, Guiyue Wang, and Fucheng Zhao. 2022. "Genome-Wide Transcriptome Analysis Revealing the Genes Related to Sugar Metabolism in Kernels of Sweet Corn" Metabolites 12, no. 12: 1254. https://doi.org/10.3390/metabo12121254
APA StyleChen, B., Feng, S., Hou, J., Zhu, Y., Bao, F., Han, H., Tan, H., Wang, G., & Zhao, F. (2022). Genome-Wide Transcriptome Analysis Revealing the Genes Related to Sugar Metabolism in Kernels of Sweet Corn. Metabolites, 12(12), 1254. https://doi.org/10.3390/metabo12121254