

Inhibition of GCN2 Reveals Synergy with Cell-Cycle Regulation and Proteostasis




Abstract
:1. Introduction
2. Materials and Methods
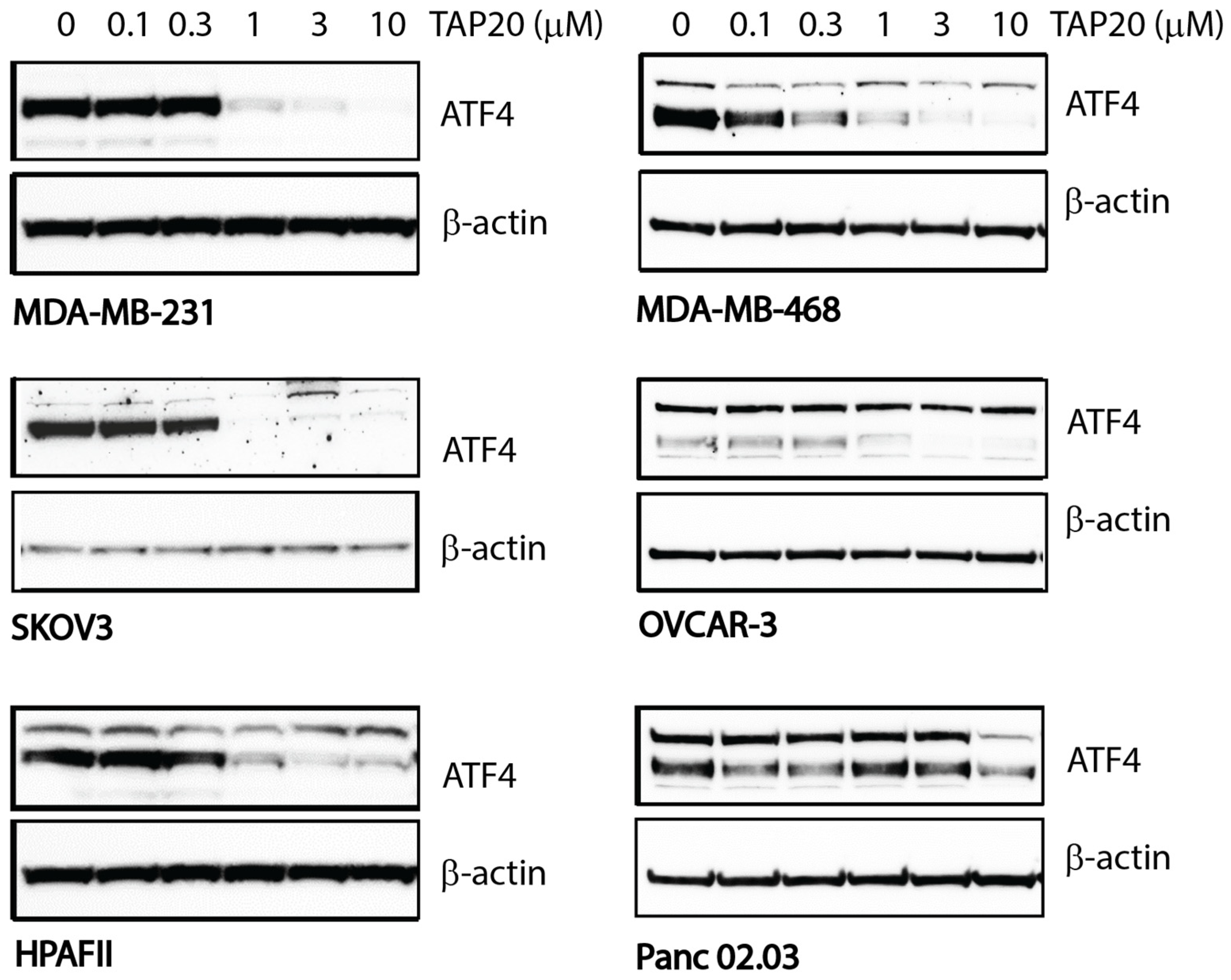
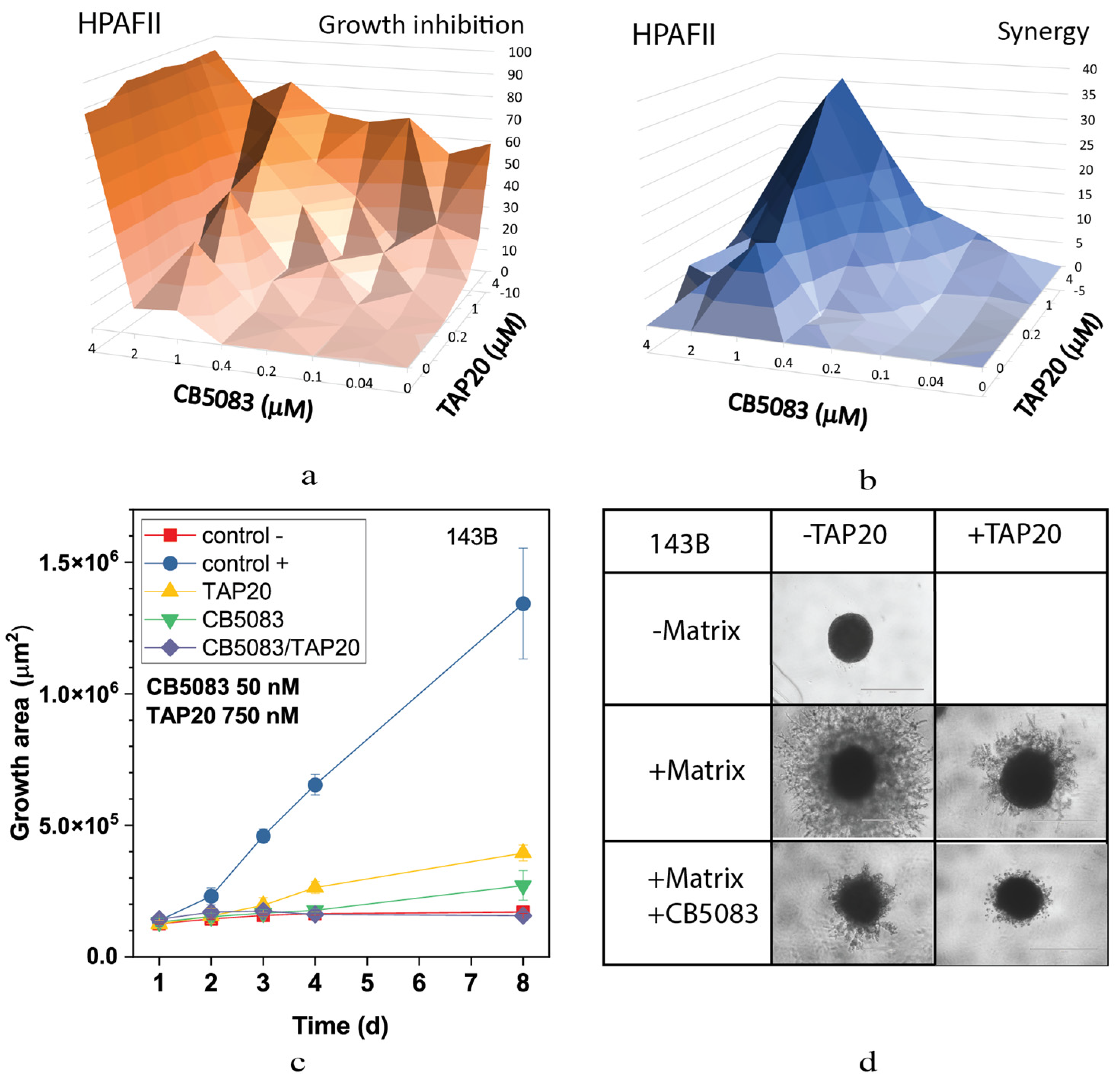
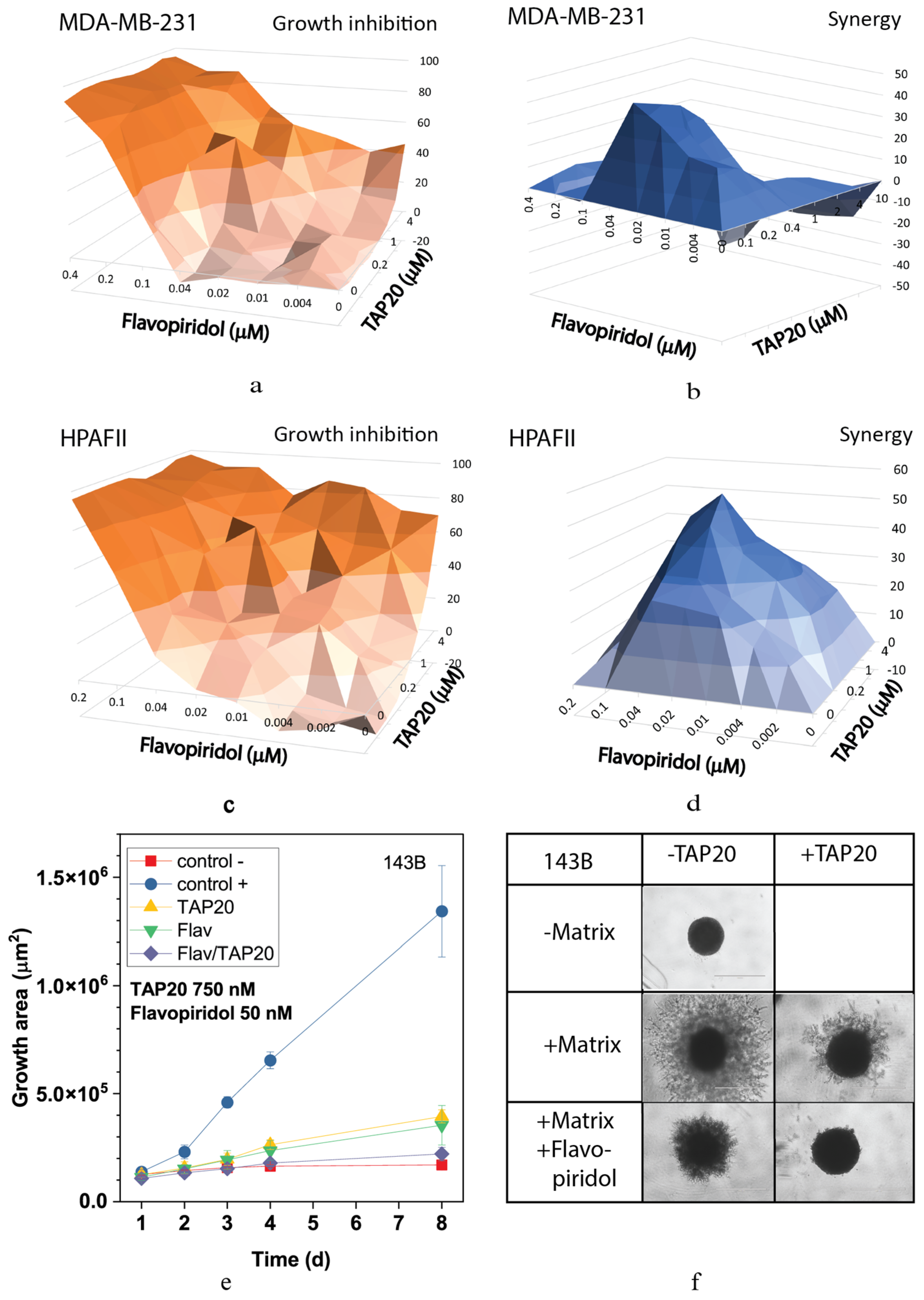
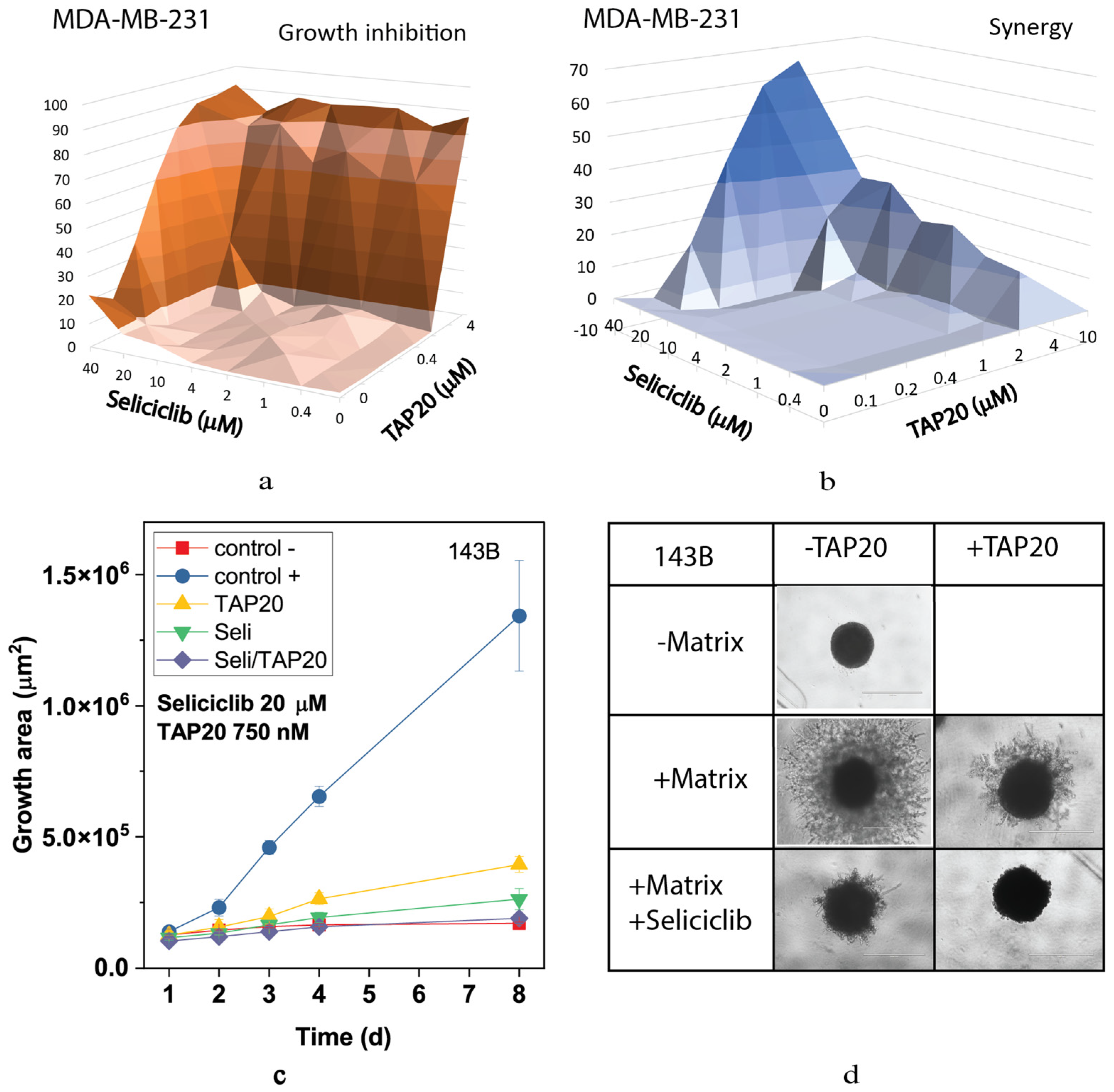
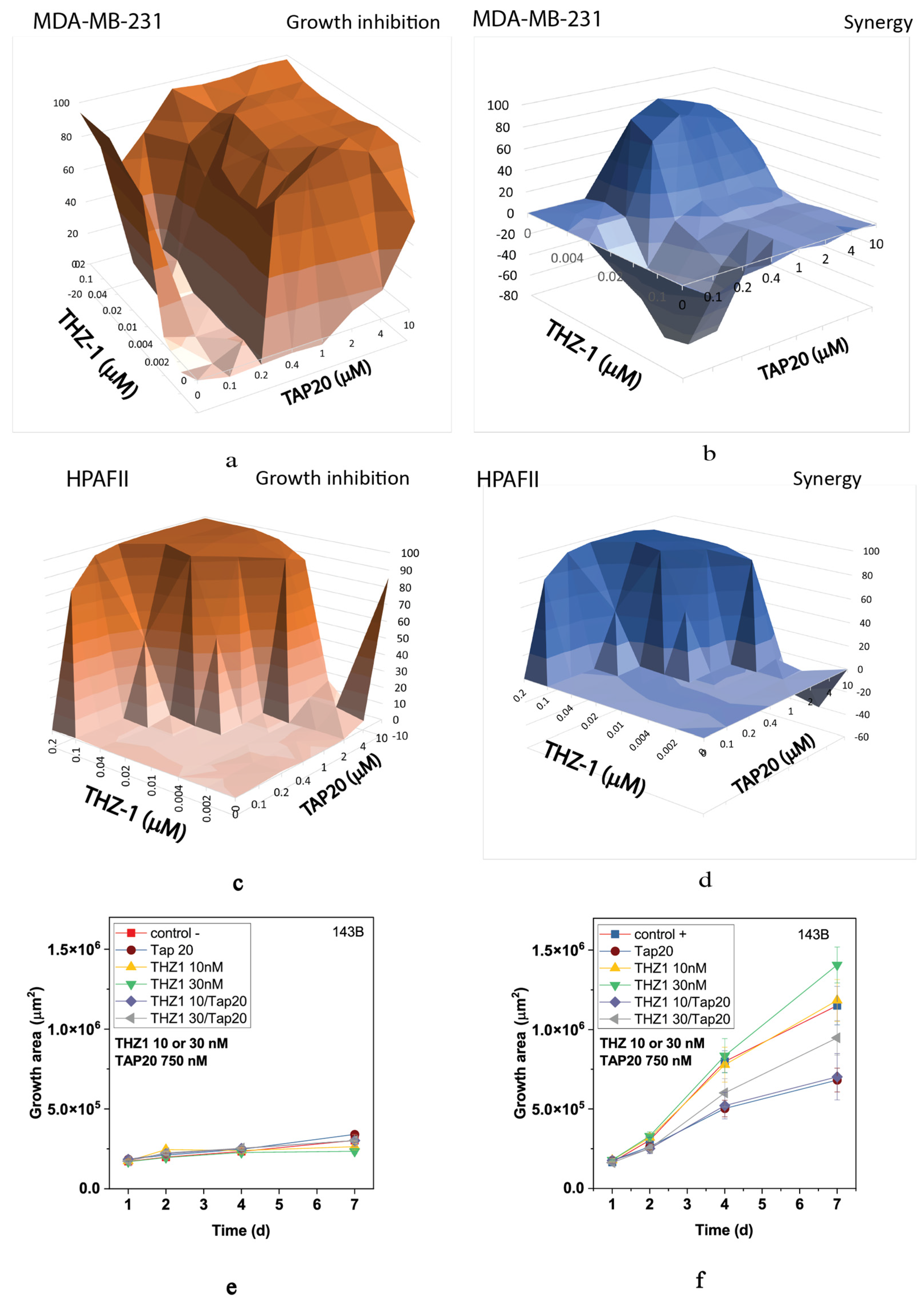
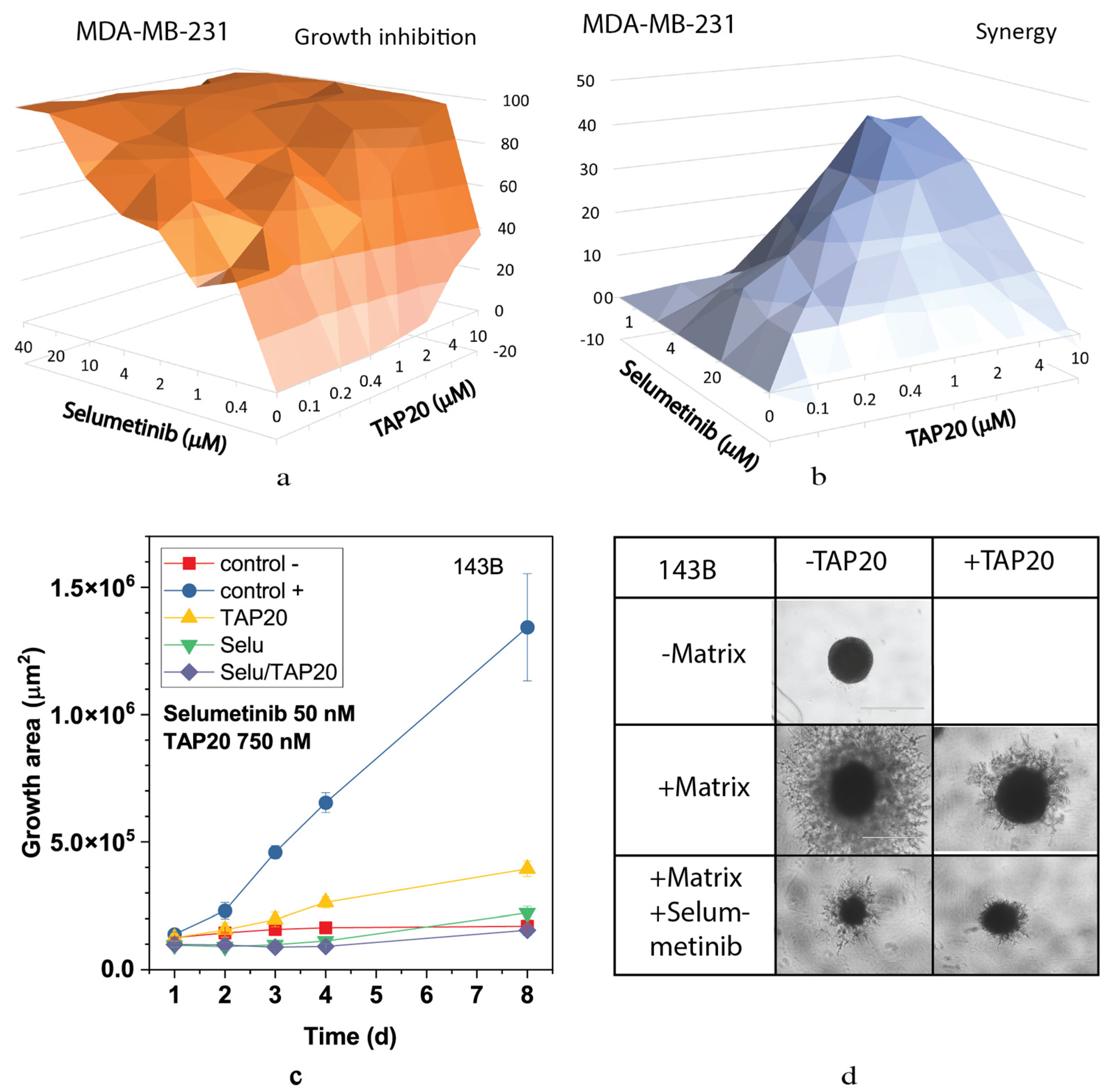
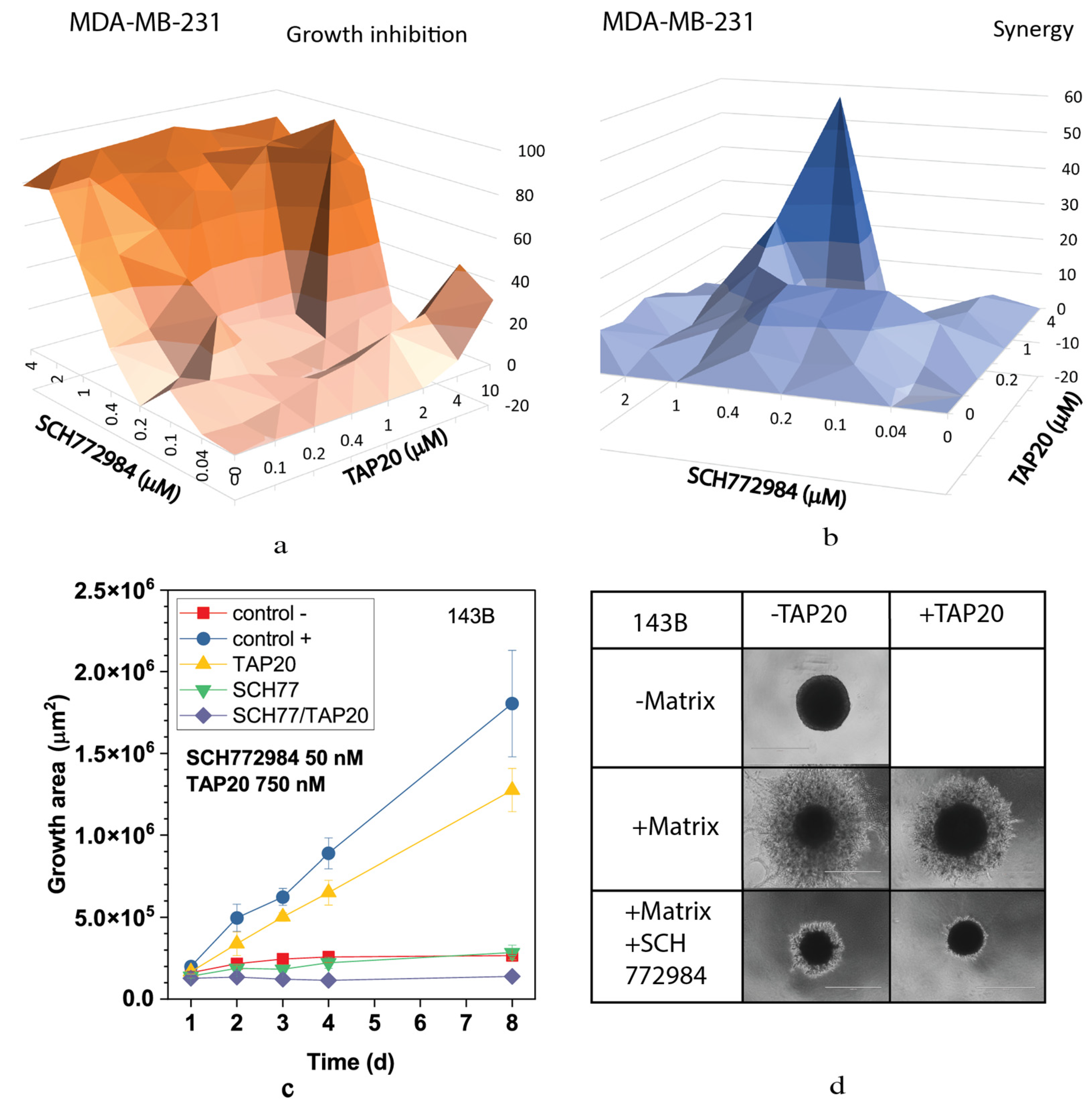
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tian, X.; Zhang, S.; Zhou, L.; Seyhan, A.A.; Hernandez Borrero, L.; Zhang, Y.; El-Deiry, W.S. Targeting the Integrated Stress Response in Cancer Therapy. Front. Pharmacol. 2021, 12, 747837. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Levin, D.H.; Petryshyn, R.; London, I.M. Characterization of double-stranded-RNA-activated kinase that phosphorylates alpha subunit of eukaryotic initiation factor 2 (eIF-2 alpha) in reticulocyte lysates. Proc. Natl. Acad. Sci. USA 1980, 77, 832–836. [Google Scholar] [CrossRef]
- Chen, J.J.; London, I.M. Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem. Sci. 1995, 20, 105–108. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Cordova, R.A.; Misra, J.; Amin, P.H.; Klunk, A.J.; Damayanti, N.P.; Carlson, K.R.; Elmendorf, A.J.; Kim, H.G.; Mirek, E.T.; Elzey, B.D.; et al. GCN2 eIF2 kinase promotes prostate cancer by maintaining amino acid homeostasis. Elife 2022, 11, e81083. [Google Scholar] [CrossRef] [PubMed]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salas Salinas, C.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.T.; Masson, G.R. GCN2: Roles in tumour development and progression. Biochem. Soc. Trans. 2022, 50, 737–745. [Google Scholar] [CrossRef]
- Jin, Y.; Saatcioglu, F. Targeting the Unfolded Protein Response in Hormone-Regulated Cancers. Trends Cancer 2020, 6, 160–171. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, R.C. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem. 2005, 280, 14189–14202. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Rahimi, F.; Broer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl. Acad. Sci. USA 2018, 115, E7776–E7785. [Google Scholar] [CrossRef]
- Okano, N.; Kawai, K.; Yamauchi, Y.; Kobayashi, T.; Naruge, D.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phaseⅠstudy of JPH203 in patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, 419. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Lane, H.A.; Breuleux, M. Optimal targeting of the mTORC1 kinase in human cancer. Curr. Opin. Cell Biol. 2009, 21, 219–229. [Google Scholar] [CrossRef]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Ables, G.P.; Hens, J.R.; Nichenametla, S.N. Methionine restriction beyond life-span extension. Ann. N. Y. Acad. Sci. 2016, 1363, 68–79. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef]
- Jaaks, P.; Coker, E.A.; Vis, D.J.; Edwards, O.; Carpenter, E.F.; Leto, S.M.; Dwane, L.; Sassi, F.; Lightfoot, H.; Barthorpe, S.; et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature 2022, 603, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kunimasa, K.; Takahashi, M.; Harada, A.; Nagasawa, I.; Osawa, M.; Sugimoto, Y.; Tomida, A. GZD824 Inhibits GCN2 and Sensitizes Cancer Cells to Amino Acid Starvation Stress. Mol. Pharmacol. 2020, 98, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, D.; Hölzemann, G.; Calderini, M.; Wegener, A.; Pöschke, O. Triazolo [4,5-d]pyrimidine Derivatives for the Treatment of Diseases Such as Cancer. International Patent WO/2014/135244, 12 September 2014. [Google Scholar]
- Broer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Broer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef] [PubMed]
- Brazeau, J.F.; Rosse, G. Triazolo [4,5-d]pyrimidine Derivatives as Inhibitors of GCN2. ACS Med. Chem. Lett. 2014, 5, 282–283. [Google Scholar] [CrossRef]
- Lines, C.L.; McGrath, M.J.; Dorwart, T.; Conn, C.S. The integrated stress response in cancer progression: A force for plasticity and resistance. Front. Oncol. 2023, 13, 1206561. [Google Scholar] [CrossRef]
- Missiaen, R.; Anderson, N.M.; Kim, L.C.; Nance, B.; Burrows, M.; Skuli, N.; Carens, M.; Riscal, R.; Steensels, A.; Li, F.; et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. 2022, 34, 1151–1167.e1157. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Tanoli, Z.; Pessia, A.; Tang, J. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genom. Proteom. Bioinform. 2022, 20, 587–596. [Google Scholar] [CrossRef]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef]
- Kim, D.G.; Lee, J.Y.; Kwon, N.H.; Fang, P.; Zhang, Q.; Wang, J.; Young, N.L.; Guo, M.; Cho, H.Y.; Mushtaq, A.U.; et al. Chemical inhibition of prometastatic lysyl-tRNA synthetase-laminin receptor interaction. Nat. Chem. Biol. 2014, 10, 29–34. [Google Scholar] [CrossRef]
- Parzych, K.; Saavedra-Garcia, P.; Valbuena, G.N.; Al-Sadah, H.A.; Robinson, M.E.; Penfold, L.; Kuzeva, D.M.; Ruiz-Tellez, A.; Loaiza, S.; Holzmann, V.; et al. The coordinated action of VCP/p97 and GCN2 regulates cancer cell metabolism and proteostasis during nutrient limitation. Oncogene 2019, 38, 3216–3231. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res. 2009, 69, 1545–1552. [Google Scholar] [CrossRef]
- Broer, A.; Fairweather, S.; Broer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharmacol. 2018, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; King, B.; Josselsohn, R.H.; Violante, S.; Macera, V.L.; Vardhana, S.A.; Cross, J.R.; Thompson, C.B. Translation in amino-acid-poor environments is limited by tRNA(Gln) charging. Elife 2020, 9, e62307. [Google Scholar] [CrossRef] [PubMed]
- Mahameed, M.; Boukeileh, S.; Obiedat, A.; Darawshi, O.; Dipta, P.; Rimon, A.; McLennan, G.; Fassler, R.; Reichmann, D.; Karni, R.; et al. Pharmacological induction of selective endoplasmic reticulum retention as a strategy for cancer therapy. Nat. Commun. 2020, 11, 1304. [Google Scholar] [CrossRef] [PubMed]
- Krokowski, D.; Han, J.; Saikia, M.; Majumder, M.; Yuan, C.L.; Guan, B.J.; Bevilacqua, E.; Bussolati, O.; Broer, S.; Arvan, P.; et al. A self-defeating anabolic program leads to beta-cell apoptosis in endoplasmic reticulum stress-induced diabetes via regulation of amino acid flux. J. Biol. Chem. 2013, 288, 17202–17213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, J.; Liu, X.; Xiao, Z.; Lei, G.; Lee, H.; Koppula, P.; Cheng, W.; Mao, C.; Zhuang, L.; et al. H2A Monoubiquitination Links Glucose Availability to Epigenetic Regulation of the Endoplasmic Reticulum Stress Response and Cancer Cell Death. Cancer Res. 2020, 80, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, I.; Kunimasa, K.; Tsukahara, S.; Tomida, A. BRAF-mutated cells activate GCN2-mediated integrated stress response as a cytoprotective mechanism in response to vemurafenib. Biochem. Biophys. Res. Commun. 2017, 482, 1491–1497. [Google Scholar] [CrossRef]
- Stretton, C.; Lipina, C.; Hyde, R.; Cwiklinski, E.; Hoffmann, T.M.; Taylor, P.M.; Hundal, H.S. CDK7 is a component of the integrated stress response regulating SNAT2 (SLC38A2)/System A adaptation in response to cellular amino acid deprivation. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 978–991. [Google Scholar] [CrossRef]
- Leung-Pineda, V.; Pan, Y.; Chen, H.; Kilberg, M.S. Induction of p21 and p27 expression by amino acid deprivation of HepG2 human hepatoma cells involves mRNA stabilization. Biochem. J. 2004, 379, 79–88. [Google Scholar] [CrossRef]
- Rzymski, T.; Harris, A.L. The unfolded protein response and integrated stress response to anoxia. Clin. Cancer Res. 2007, 13, 2537–2540. [Google Scholar] [CrossRef]
- Luo, L.; Guan, X.; Begum, G.; Ding, D.; Gayden, J.; Hasan, M.N.; Fiesler, V.M.; Dodelson, J.; Kohanbash, G.; Hu, B.; et al. Blockade of Cell Volume Regulatory Protein NKCC1 Increases TMZ-Induced Glioma Apoptosis and Reduces Astrogliosis. Mol. Cancer Ther. 2020, 19, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D.; Ma, J.; Grubbs, E.G.; Gagel, R.F.; Bagheri-Yarmand, R. ATF4 loss of heterozygosity is associated with poor overall survival in medullary thyroid carcinoma. Am. J. Cancer Res. 2021, 11, 3227–3239. [Google Scholar] [PubMed]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef]
- Wengrod, J.; Wang, D.; Weiss, S.; Zhong, H.; Osman, I.; Gardner, L.B. Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci. Signal 2015, 8, ra27. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutte, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic Disruption of the Multifunctional CD98/LAT1 Complex Demonstrates the Key Role of Essential Amino Acid Transport in the Control of mTORC1 and Tumor Growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Meulenbeld, H.J.; Mathijssen, R.H.; Verweij, J.; de Wit, R.; de Jonge, M.J. Danusertib, an aurora kinase inhibitor. Expert. Opin. Investig. Drugs 2012, 21, 383–393. [Google Scholar] [CrossRef]
- Valles-Marti, A.; Mantini, G.; Manoukian, P.; Waasdorp, C.; Sarasqueta, A.F.; de Goeij-de Haas, R.R.; Henneman, A.A.; Piersma, S.R.; Pham, T.V.; Knol, J.C.; et al. Phosphoproteomics guides effective low-dose drug combinations against pancreatic ductal adenocarcinoma. Cell Rep. 2023, 42, 112581. [Google Scholar] [CrossRef]
- Pecoraro, C.; Carbone, D.; Cascioferro, S.M.; Parrino, B.; Diana, P. Multi or Single-Kinase Inhibitors to Counteract Drug Resistance in Cancer: What is New? Curr. Med. Chem. 2023, 30, 776–782. [Google Scholar] [CrossRef]
- Finicle, B.T.; Jayashankar, V.; Edinger, A.L. Nutrient scavenging in cancer. Nat. Rev. Cancer 2018, 18, 619–633. [Google Scholar] [CrossRef]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front. Oncol. 2018, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Nofal, M.; Wang, T.; Yang, L.; Jankowski, C.S.R.; Hsin-Jung Li, S.; Han, S.; Parsons, L.; Frese, A.N.; Gitai, Z.; Anthony, T.G.; et al. GCN2 adapts protein synthesis to scavenging-dependent growth. Cell Syst. 2022, 13, 158–172.e159. [Google Scholar] [CrossRef] [PubMed]
- Broer, S.; Broer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017, 474, 1935–1963. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I.; Bishop, A.J.; Motoyama, N.; Cao, L.; Finkel, T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef]
- Nigg, E.A. Cyclin-dependent kinase 7: At the cross-roads of transcription, DNA repair and cell cycle control? Curr. Opin. Cell Biol. 1996, 8, 312–317. [Google Scholar] [CrossRef]
- Fisher, R.P. Cdk7: A kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2019, 10, 47–56. [Google Scholar] [CrossRef]
- Gaccioli, F.; Huang, C.C.; Wang, C.; Bevilacqua, E.; Franchi-Gazzola, R.; Gazzola, G.C.; Bussolati, O.; Snider, M.D.; Hatzoglou, M. Amino acid starvation induces the SNAT2 neutral amino acid transporter by a mechanism that involves eukaryotic initiation factor 2alpha phosphorylation and cap-independent translation. J. Biol. Chem. 2006, 281, 17929–17940. [Google Scholar] [CrossRef] [PubMed]
- Kartha, N.; Gianopulos, J.E.; Schrank, Z.; Cavender, S.M.; Dobersch, S.; Kynnap, B.D.; Wallace-Povirk, A.; Wladyka, C.L.; Santana, J.F.; Kim, J.C.; et al. Sirtuin 6 is required for the integrated stress response and resistance to inhibition of transcriptional cyclin-dependent kinases. Sci. Transl. Med. 2023, 15, eabn9674. [Google Scholar] [CrossRef] [PubMed]
- Piecyk, M.; Triki, M.; Laval, P.A.; Duret, C.; Fauvre, J.; Cussonneau, L.; Machon, C.; Guitton, J.; Rama, N.; Gibert, B.; et al. The stress sensor GCN2 differentially controls ribosome biogenesis in colon cancer according to the nutritional context. Mol. Oncol. 2023. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Benson, C.; White, J.; De Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef]
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How proteolysis drives the cell cycle. Science 1996, 274, 1652–1659. [Google Scholar] [CrossRef]
- Cormerais, Y.; Vucetic, M.; Parks, S.K.; Pouyssegur, J. Amino Acid Transporters Are a Vital Focal Point in the Control of mTORC1 Signaling and Cancer. Int. J. Mol. Sci. 2020, 22, 23. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Thiaville, M.M.; Pan, Y.X.; Gjymishka, A.; Zhong, C.; Kaufman, R.J.; Kilberg, M.S. MEK signaling is required for phosphorylation of eIF2alpha following amino acid limitation of HepG2 human hepatoma cells. J. Biol. Chem. 2008, 283, 10848–10857. [Google Scholar] [CrossRef]
- Franchi-Gazzola, R.; Visigalli, R.; Bussolati, O.; Dall’Asta, V.; Gazzola, G.C. Adaptive increase of amino acid transport system A requires ERK1/2 activation. J. Biol. Chem. 1999, 274, 28922–28928. [Google Scholar] [CrossRef]
- Doherty, M.K.; Tam, V.C.; McNamara, M.G.; Jang, R.; Hedley, D.; Chen, E.; Dhani, N.; Tang, P.; Sim, H.W.; O’Kane, G.M.; et al. Randomised, Phase II study of selumetinib, an oral inhibitor of MEK, in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer. Br. J. Cancer 2022, 127, 1473–1478. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | IC50 |
|---|---|---|
| Agerafenib | BRAFV600E | 14 nM |
| AMG PERK44 | PERK | 6 nM |
| AZD3965 | MCT1 | 1.6 nM |
| Bay876 | GLUT1 | 2 nM |
| Bortezomib | 20S proteasome | 0.6 nM |
| Bumetanide | NKCC1 | 680 nM |
| CB-839 | Glutaminase 1 | 25 nM |
| CB-5083 | p97AAA ATPase | 11 nM |
| Cetuximab | EGFR | 0.2 nM |
| Danusertib | Aurora-kinases | 13–79 nM |
| GSK2837808A | LDH-A | 2.6 nM |
| GZD824 | Bcr-Abl | 0.32 nM |
| Flavopiridol | CDK (non-specific) | 6–300 nM |
| 5-Fluorouracil | Thymidylate synthase | Irr 1 |
| Halofuginone | Prolyl-tRNA synthetase | 18 nM |
| NCT-503 | PHGDH | 2500 nM |
| PLX8394 | BRAF/BRAFV600E | 14 nM/5 nM |
| Rapamycin | FKBP12 | 0.1 nM |
| SCH772984 | ERK1/2 | 4 nM/1 nM |
| Seliciclib | CDK (non-specific) | 200–800 nM |
| Selumetinib | MEK1/2 | 14 nM |
| Thapsigargin | SERCA | 0.4 nM |
| THZ-1 | CDK7 | Irr 1 |
| V-9302 | SNAT2/LAT1 | n.d. |
| YH16899 | KRS-67LR interaction | 8600 nM |
| Compound | MDA-MB-231 | MDA-MB-468 | HPAFII | Panc02.03 | SKOV3 | OVCAR3 |
|---|---|---|---|---|---|---|
| Agerafenib | 4.3 | 4.3 | 1.6 | 3.7 | 4.7 | 6.5 |
| AMG PERK44 | >10 | >10 | 9 | >10 | >10 | >10 |
| AZD3965 | >10 | >10 | >10 | >10 | >10 | >10 |
| Bay876 | 10 | 10 | 0.25 | 0.15 | 1.4 | 0.16 |
| Bortezomib | 0.002 | 0.003 | 0.003 | 0.004 | 0.004 | 0.002 |
| Bumetanide | n.i. | n.d. | n.i. | n.d. | n.i. | n.d. |
| CB-839 | 10 | n.d. | >10 | n.d. | >10 | n.d. |
| CB-5083 | 0.76 | 0.36 | 0.19 | 0.12 | 1.2 | 0.36 |
| Cetuximab | >10 | n.d. | >10 | n.d. | >10 | n.d. |
| Danusertib | <0.1 | <0.1 | 0.8 | 0.36 | 2.2 | <0.1 |
| GSK2837808A | >10 | n.d. | >10 | n.d. | >10 | n.d. |
| GZD824 | 1 | 1.3 | <0.1 | 0.31 | 1.6 | 1.8 |
| Flavopiridol | 0.1 | 0.04 | <0.1 | 4 | 0.22 | 1.7 |
| 5-Fluorouracil | >10 | >10 | >10 | 8.9 | >10 | 1.7 |
| Halofuginone | 0.032 | 0.054 | 0.038 | 0.057 | 0.019 | 0.058 |
| NCT-503 | >10 | >10 | >10 | >10 | >10 | >10 |
| PLX8394 | 10 | 10 | 4.5 | 10 | 6 | 3.6 |
| Rapamycin | >10 | n.d. | >10 | n.d. | >10 | n.d. |
| SCH772984 | 1 | 1 | 0.45 | 0.4 | 8.4 | 5 |
| Seliciclib | >10 | >10 | 9.5 | 6.3 | >10 | 7.4 |
| Selumetinib | 10 | 6.3 | <0.1 | 0.88 | 10 | 1.4 |
| Thapsigargin | <0.1 | 0.005 | <0.1 | 0.004 | 0.52 | <0.003 |
| V-9302 | 1.1 | 1.1 | 1.1 | 2 | 2.6 | 1.1 |
| YH16899 | >10 | n.d. | >10 | n.d. | >10 | n.d |
| Compound | MDA-MB-231 | MDA-MB-468 | HPAFII | Panc02.03 | SKOV3 | OVCAR3 |
| Agerafenib | 3 | 3 | 1 | 0.3 | 3 | 3 |
| AMG PERK44 | 10 | 10 | 10 | 10 | 10 | 10 |
| AZD3965 | 10 | 10 | 10 | 10 | 10 | 3 |
| Bay876 | 10 | 3 | 0.1 | 0.03 | 1 | 0.1 |
| Bortezomib | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| Bumetanide | 10 | 10 | 10 | 10 | 10 | 10 |
| CB-839 | 10 | 10 | 10 | 10 | 10 | 10 |
| CB-5083 | 1 | 0.3 | 0.1 | 0.1 | 1 | 0.3 |
| Cetuximab | 0.033 | 0.033 | 0.033 | 0.033 | 0.033 | 0.033 |
| Danusertib | 0.01 | 0.03 | 0.3 | 0.1 | 1 | 0.01 |
| GSK2837808A | 10 | 10 | 10 | 10 | 10 | 10 |
| GZD824 | 0.3 | 0.3 | 0.01 | 1 | 0.1 | 0.1 |
| Flavopiridol | 0.1 | 0.03 | 0.03 | 3 | 0.1 | 1 |
| 5-Fluorouracil | 10 | 3 | 3 | 3 | 10 | 1 |
| Halofuginone | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| NCT-503 | 10 | 10 | 10 | 10 | 10 | 10 |
| PLX8394 | 10 | 3 | 3 | 3 | 3 | 1 |
| Rapamycin | 10 | 10 | 10 | 10 | 10 | 10 |
| SCH772984 | 1 | 0.3 | 0.3 | 0.1 | 3 | 0.1 |
| Seliciclib | 10 | 3 | 3 | 3 | 10 | 3 |
| Selumetinib | 10 | 3 | 0.03 | 0.3 | 10 | 0.3 |
| Thapsigargin | 0.01 | 0.001 | 0.003 | 0.001 | 0.1 | 0.001 |
| V-9302 | 1 | 1 | 1 | 1 | 1 | 1 |
| YH16899 | 10 | 10 | 10 | 10 | 10 | 10 |
| Compound | MDA-MB-231 | MDA-MB-468 | HPAFII | Panc02.03 | SKOV3 | OVCAR3 |
|---|---|---|---|---|---|---|
| Agerafenib | 1.21 | 0.96 | 0.97 | 1.07 | 1.49 | 1.57 |
| AMG PERK44 | 0.88 | 0.94 | 0.66 | 0.97 | 1.07 | 1.3 |
| AZD3965 | 0.77 | 0.79 | 0.76 | 1.05 | 0.96 | 0.84 |
| Bay876 | 0.97 | 0.63 | 0.73 | 0.73 | 0.87 | 0.72 |
| Bortezomib | 0.79 | 0.82 | 0.62 | 0.83 | 0.89 | 0.91 |
| Bumetanide | 0.83 | 0.81 | 0.86 | 0.99 | 0.97 | 0.93 |
| CB-839 | 0.86 | 0.9 | 0.89 | 1.03 | 0.97 | 0.97 |
| CB-5083 | 0.85 | 0.89 | 0.51 | 0.96 | 1.17 | 0.81 |
| Cetuximab | 1.11 | 0.77 | 1.06 | 0.97 | 0.99 | 0.91 |
| Danusertib | 0.85 | 0.65 | 0.23 | 0.67 | 0.65 | 1.53 |
| GSK2837808A | 0.85 | 0.87 | 0.78 | 1.03 | 0.99 | 0.99 |
| GZD824 | 0.82 | 0.86 | 0.87 | 0.91 | 1.5 | 1.1 |
| Flavopiridol | 0.24 | 0.96 | 0.4 | 1.06 | 0.8 | 0.56 |
| 5-Fluorouracil | 0.7 | 0.95 | 0.71 | 0.87 | 0.94 | 0.51 |
| Halofuginone | 0.78 | 0.88 | 0.53 | 0.82 | 0.73 | 0.93 |
| NCT-503 | 0.68 | 0.88 | 0.74 | 0.86 | 0.95 | 1.11 |
| PLX8394 | 0.91 | 0.6 | 0.88 | 0.88 | 1.16 | 0.97 |
| Rapamycin | 1.29 | 1.05 | 1.56 | 0.97 | 1.57 | 2.38 |
| SCH772984 | 0.23 | 1.4 | 0.29 | 0.86 | 0.53 | 0.88 |
| Seliciclib | 0.47 | 0.85 | 0.51 | 0.96 | 0.87 | 0.88 |
| Selumetinib | 0.3 | 0.63 | 1.21 | 0.7 | 0.71 | 0.63 |
| Thapsigargin | 0.87 | 0.95 | 0.57 | 1.01 | 0.69 | 0.87 |
| THZ-1 | 0.71 | n.d. | 0.12 | n.d. | 0.4 | n.d. |
| V-9302 | 0.63 | 0.96 | 0.44 | 0.84 | 0.93 | 0.77 |
| YH16899 | 0.9 | 0.75 | 0.55 | 0.94 | 0.93 | 0.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauthier-Coles, G.; Rahimi, F.; Bröer, A.; Bröer, S. Inhibition of GCN2 Reveals Synergy with Cell-Cycle Regulation and Proteostasis. Metabolites 2023, 13, 1064. https://doi.org/10.3390/metabo13101064
Gauthier-Coles G, Rahimi F, Bröer A, Bröer S. Inhibition of GCN2 Reveals Synergy with Cell-Cycle Regulation and Proteostasis. Metabolites. 2023; 13(10):1064. https://doi.org/10.3390/metabo13101064
Chicago/Turabian StyleGauthier-Coles, Gregory, Farid Rahimi, Angelika Bröer, and Stefan Bröer. 2023. "Inhibition of GCN2 Reveals Synergy with Cell-Cycle Regulation and Proteostasis" Metabolites 13, no. 10: 1064. https://doi.org/10.3390/metabo13101064
APA StyleGauthier-Coles, G., Rahimi, F., Bröer, A., & Bröer, S. (2023). Inhibition of GCN2 Reveals Synergy with Cell-Cycle Regulation and Proteostasis. Metabolites, 13(10), 1064. https://doi.org/10.3390/metabo13101064