
Metabolic Reprogramming of Castration-Resistant Prostate Cancer Cells as a Response to Chemotherapy

 ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
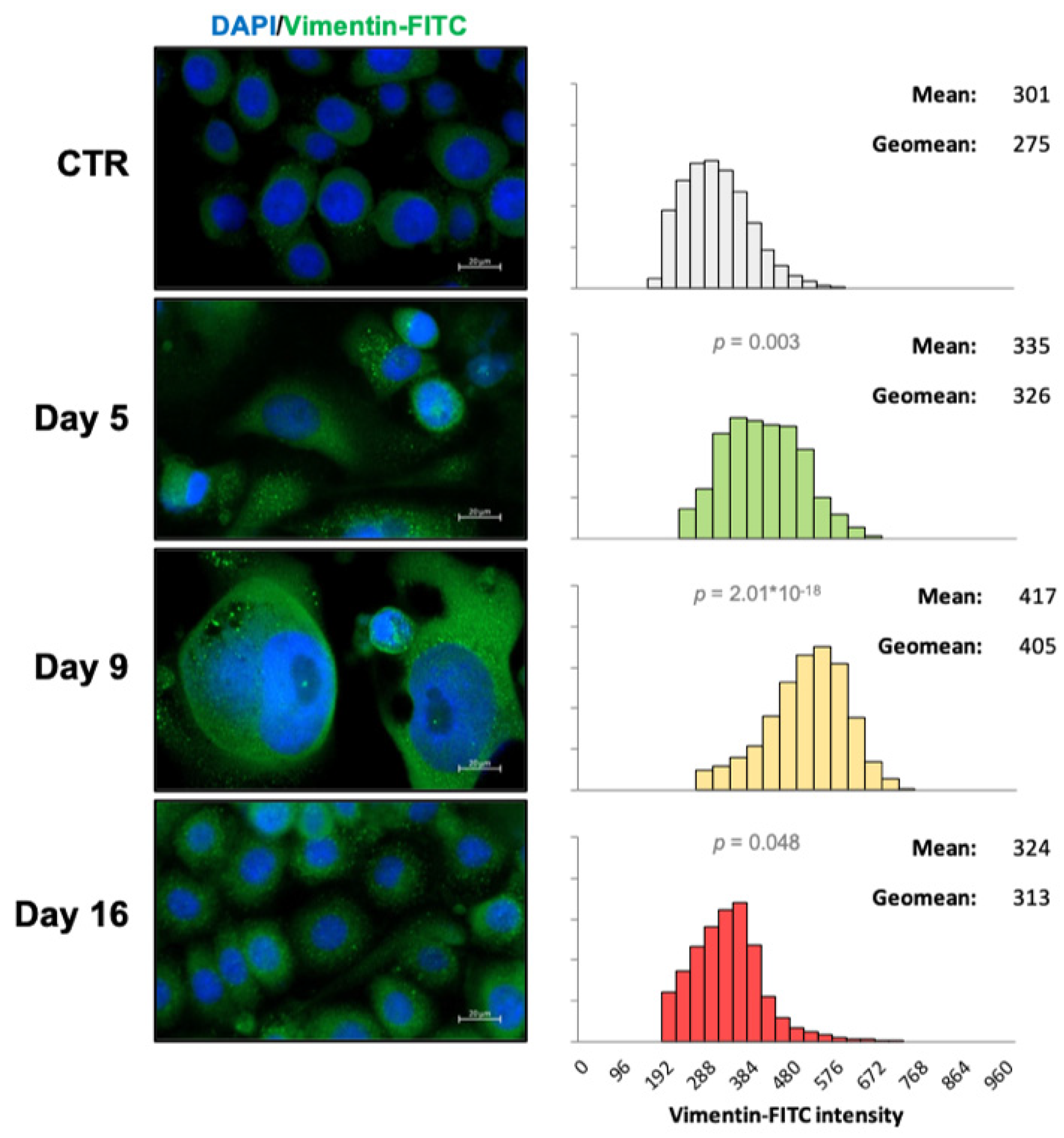
2.2. Vimentin Staining and Analysis
2.3. Samples for NMR Analysis
2.4. 1H-NMR Spectroscopy
2.5. Spectral Deconvolution
2.6. Measurement of the Metabolite Exchange Rates
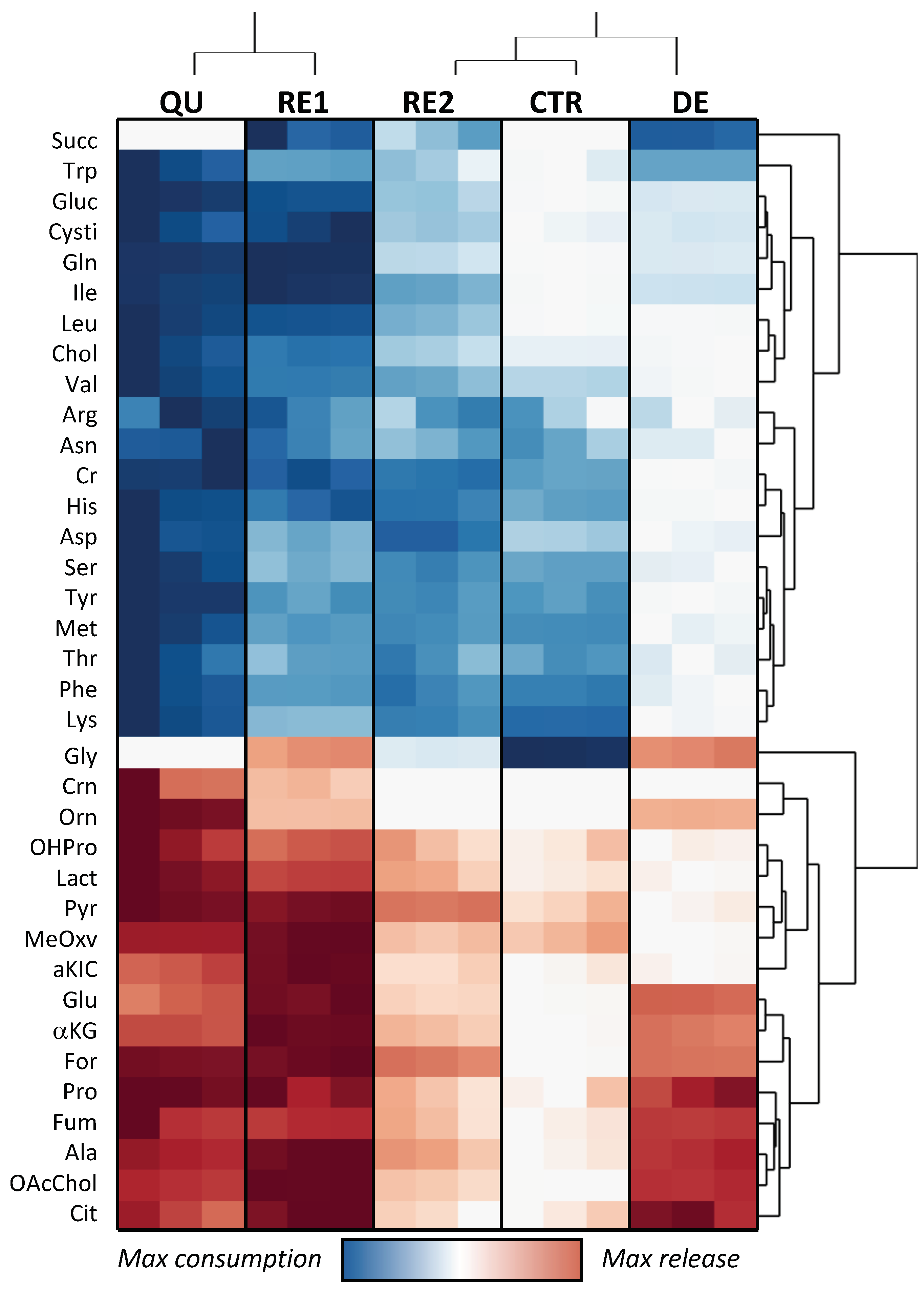
Scaling of the Variation Matrix to Obtain a Heatmap
2.7. Data Analysis
3. Results
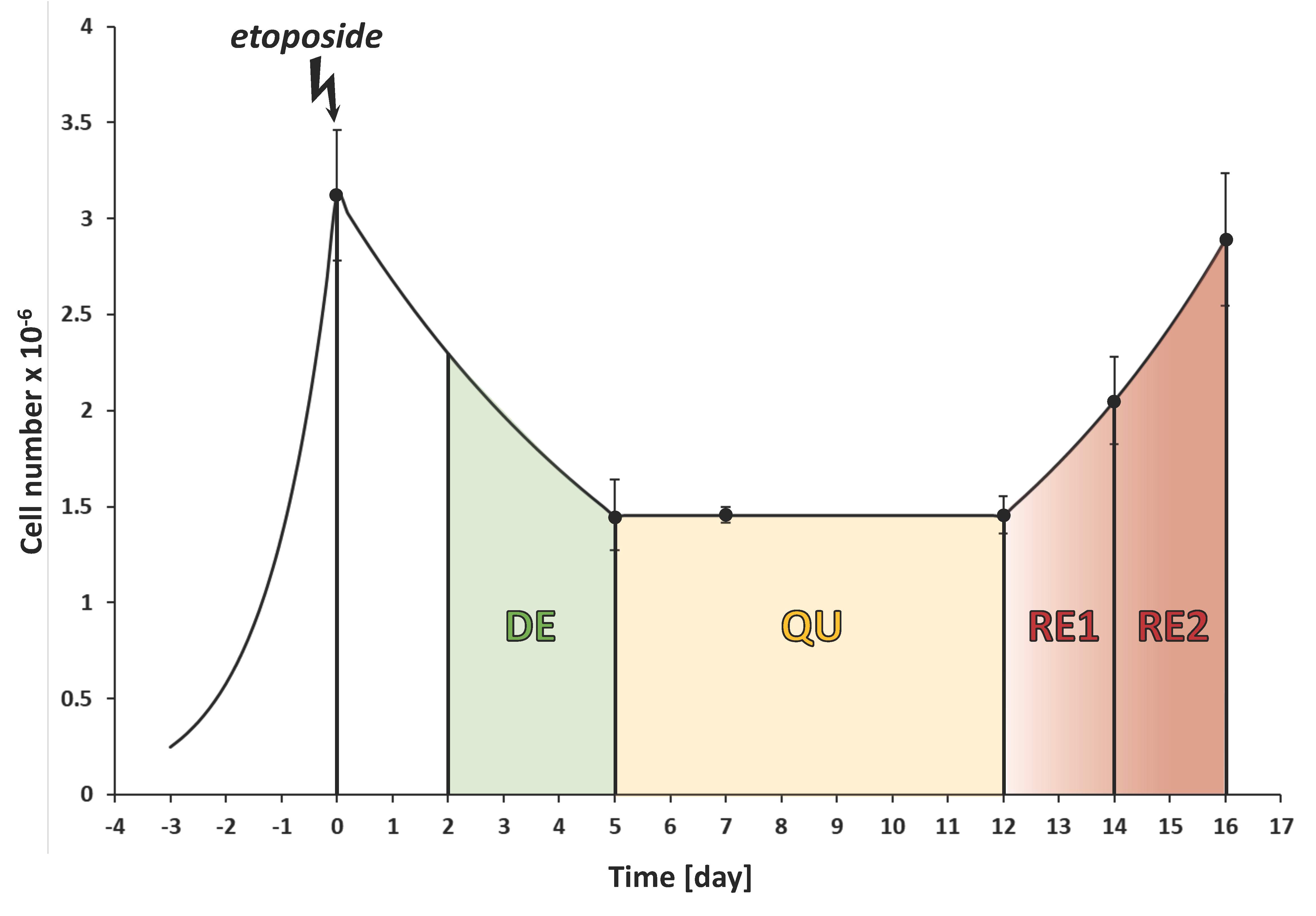
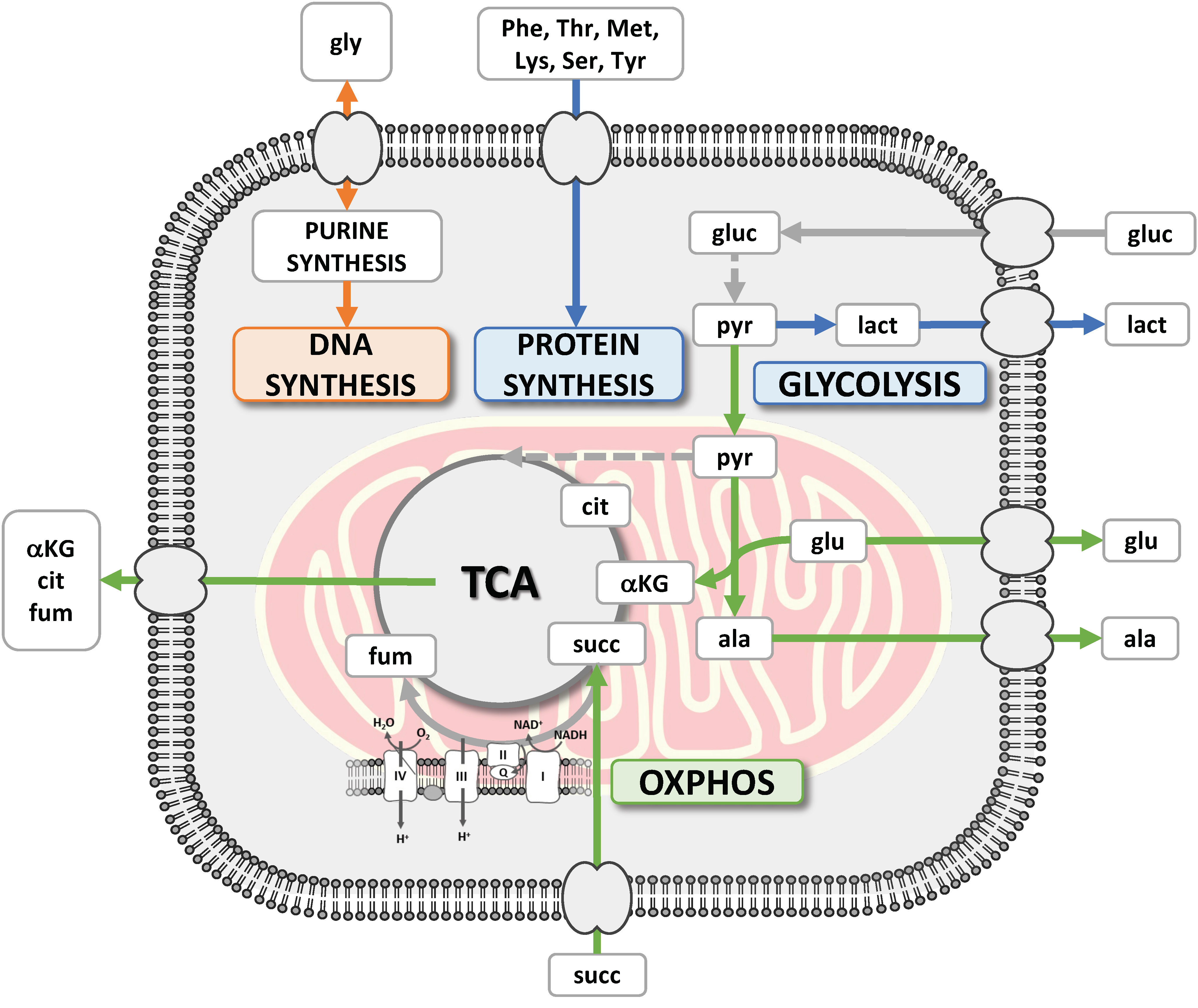
3.1. The CRAC Experiment to Study the Cell Metabolic Changes
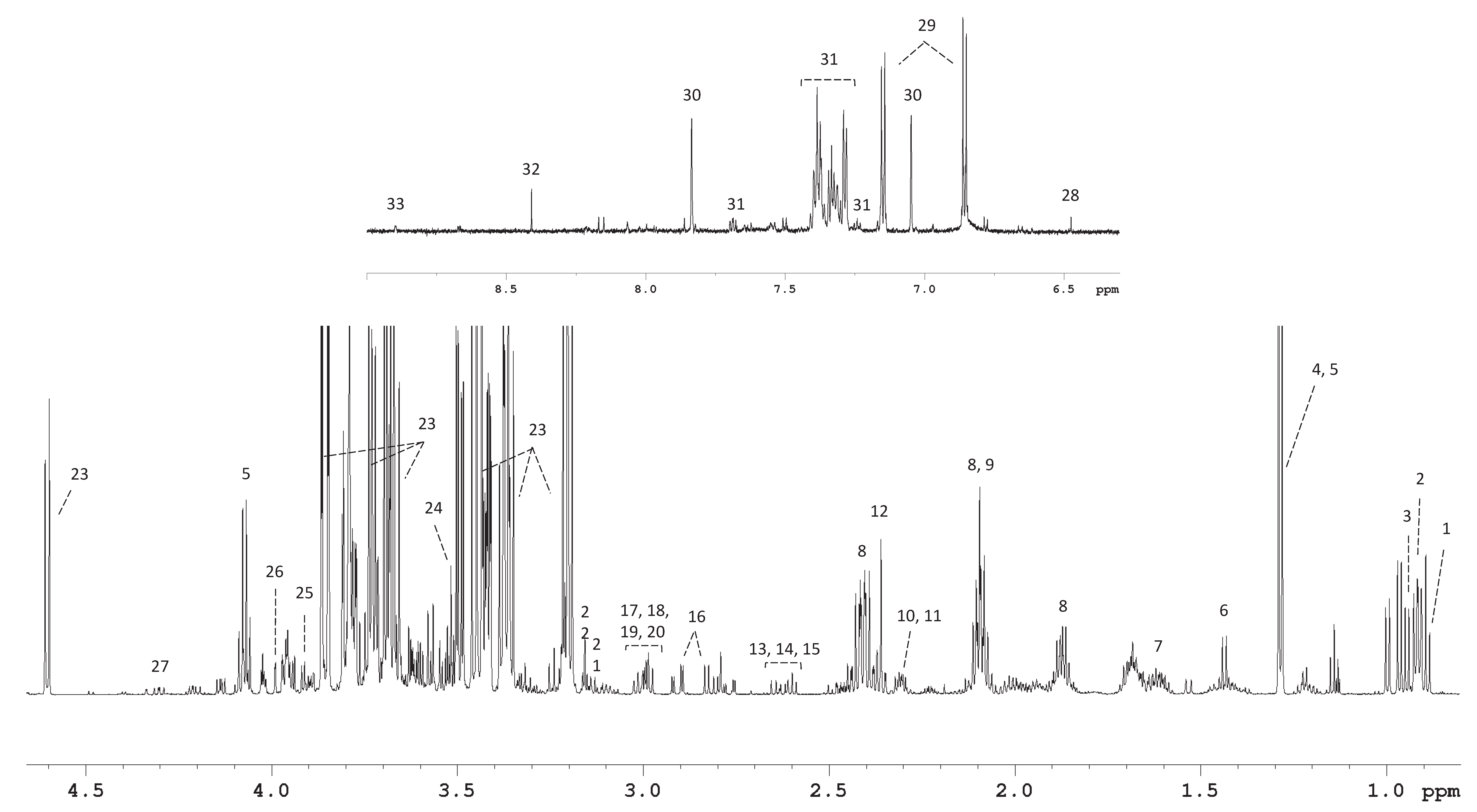
3.2. 1H NMR Spectroscopy Provided Metabolite Concentrations in Culture Medium Samples
3.3. Etoposide Exposure Alters PC3 Metabolic Exchange Rates
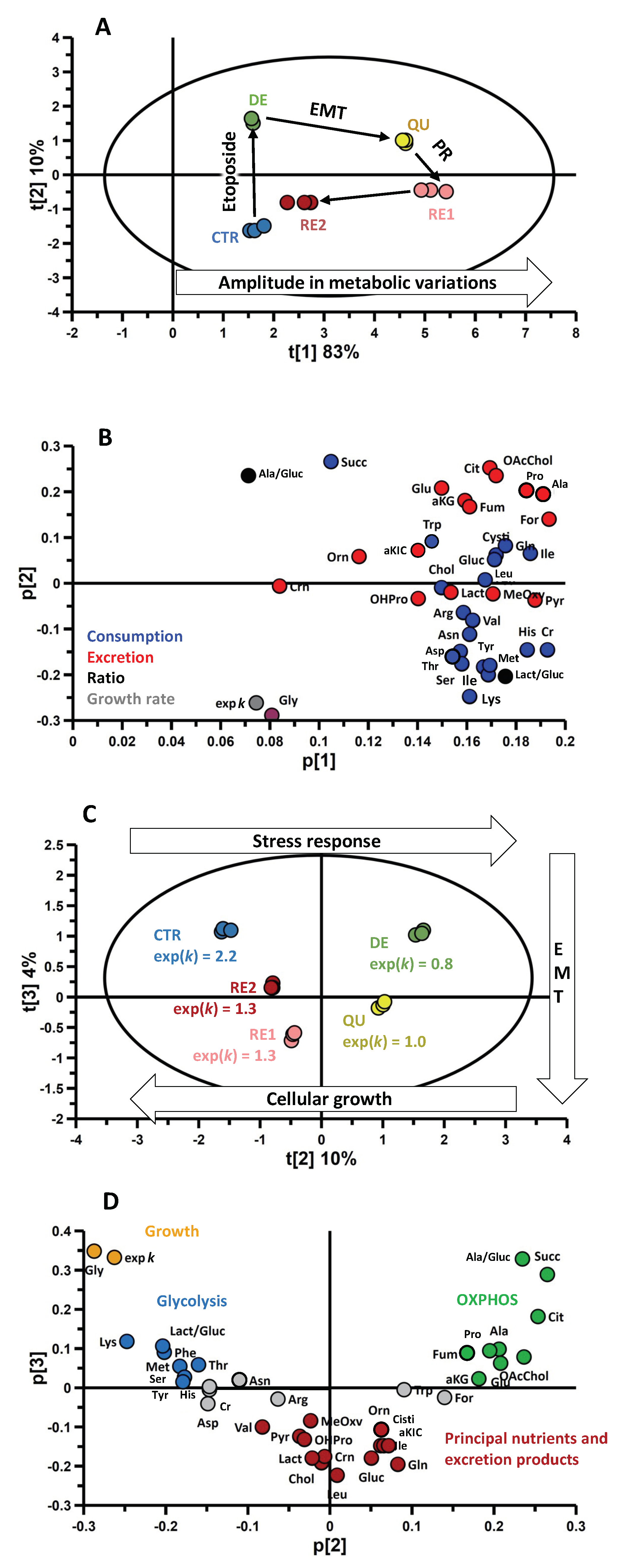
3.4. Omics Correlations Reveal Different Metabolic Regulations during Successive CRAC Phases
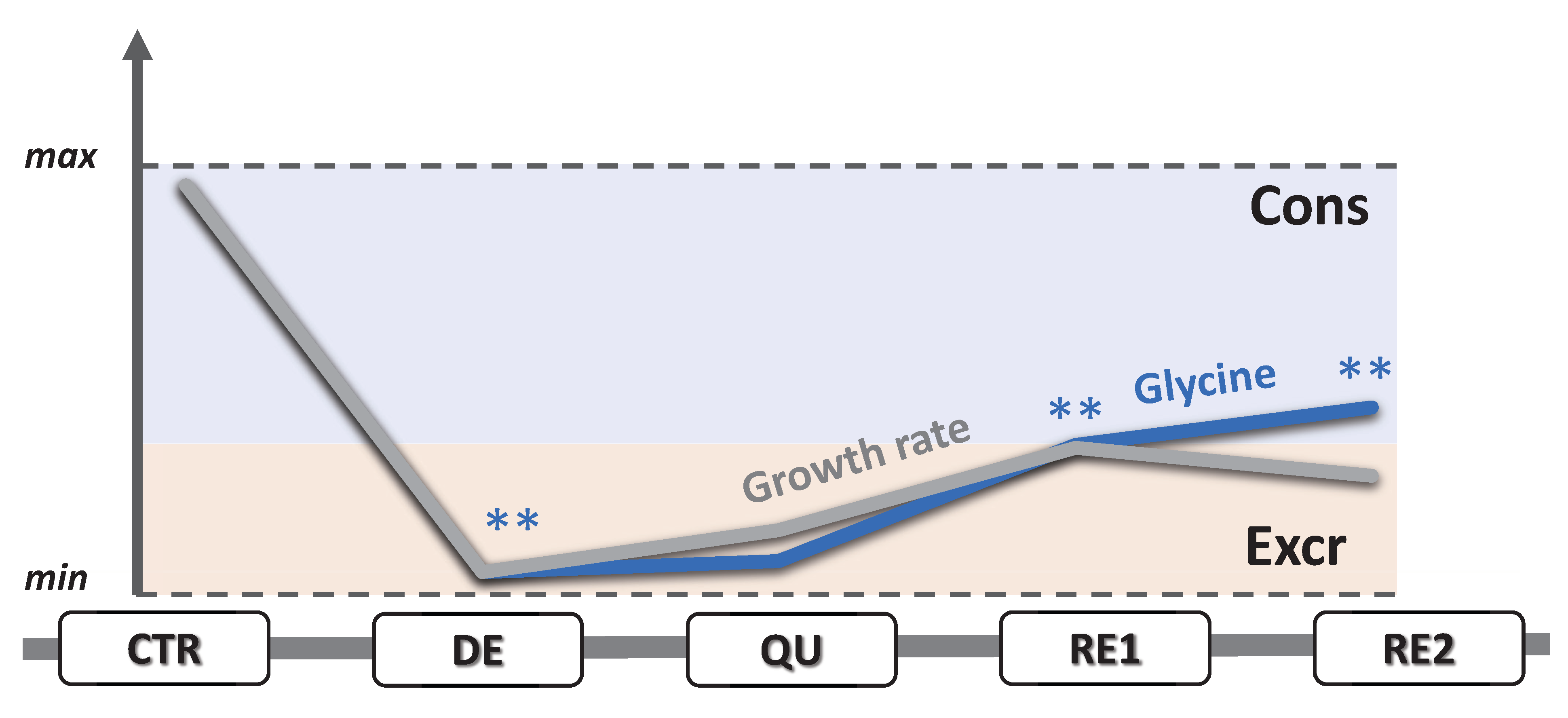
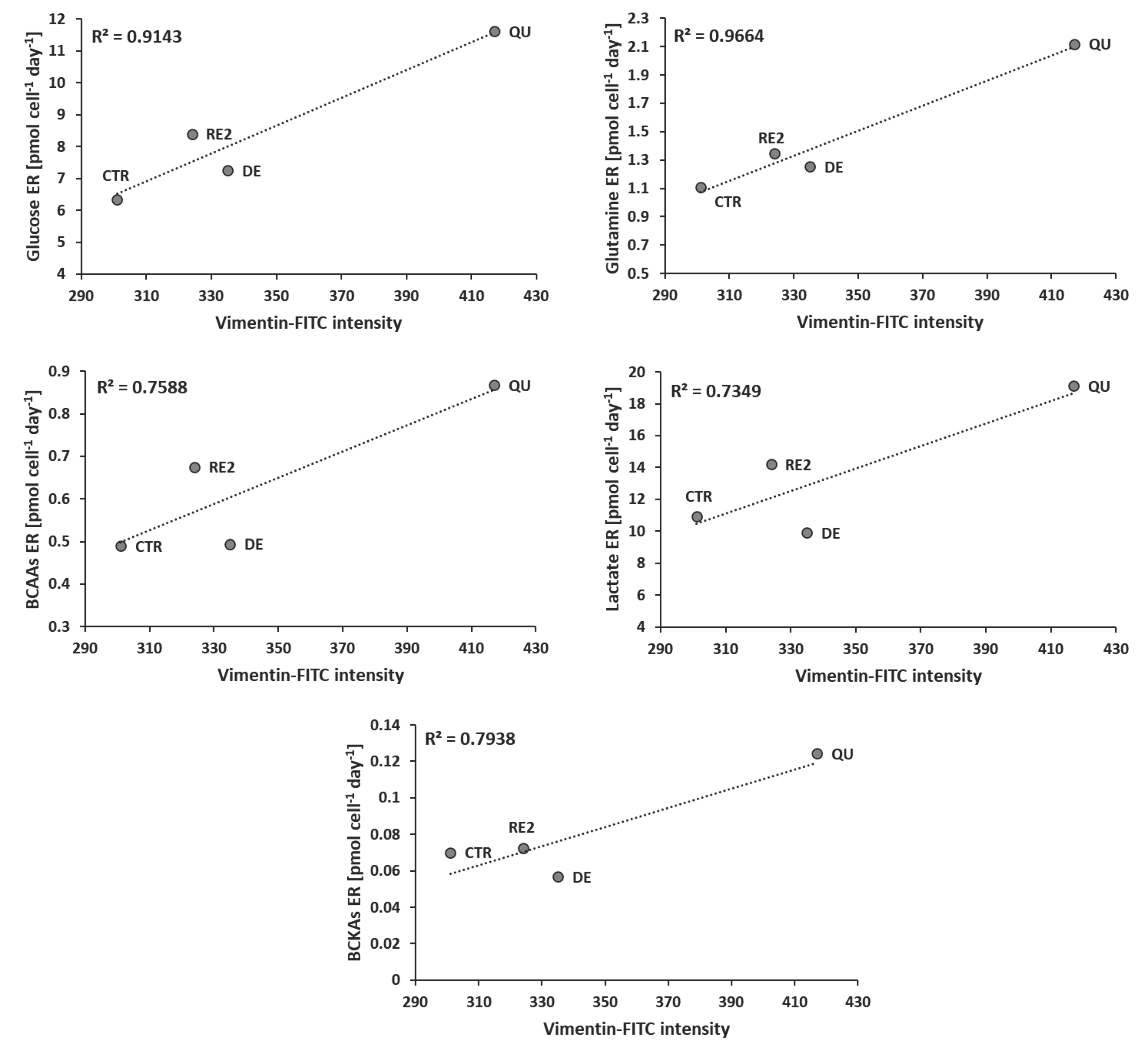
3.5. EMT Correlates with Higher Daily Consumption of the Principal Nutrients
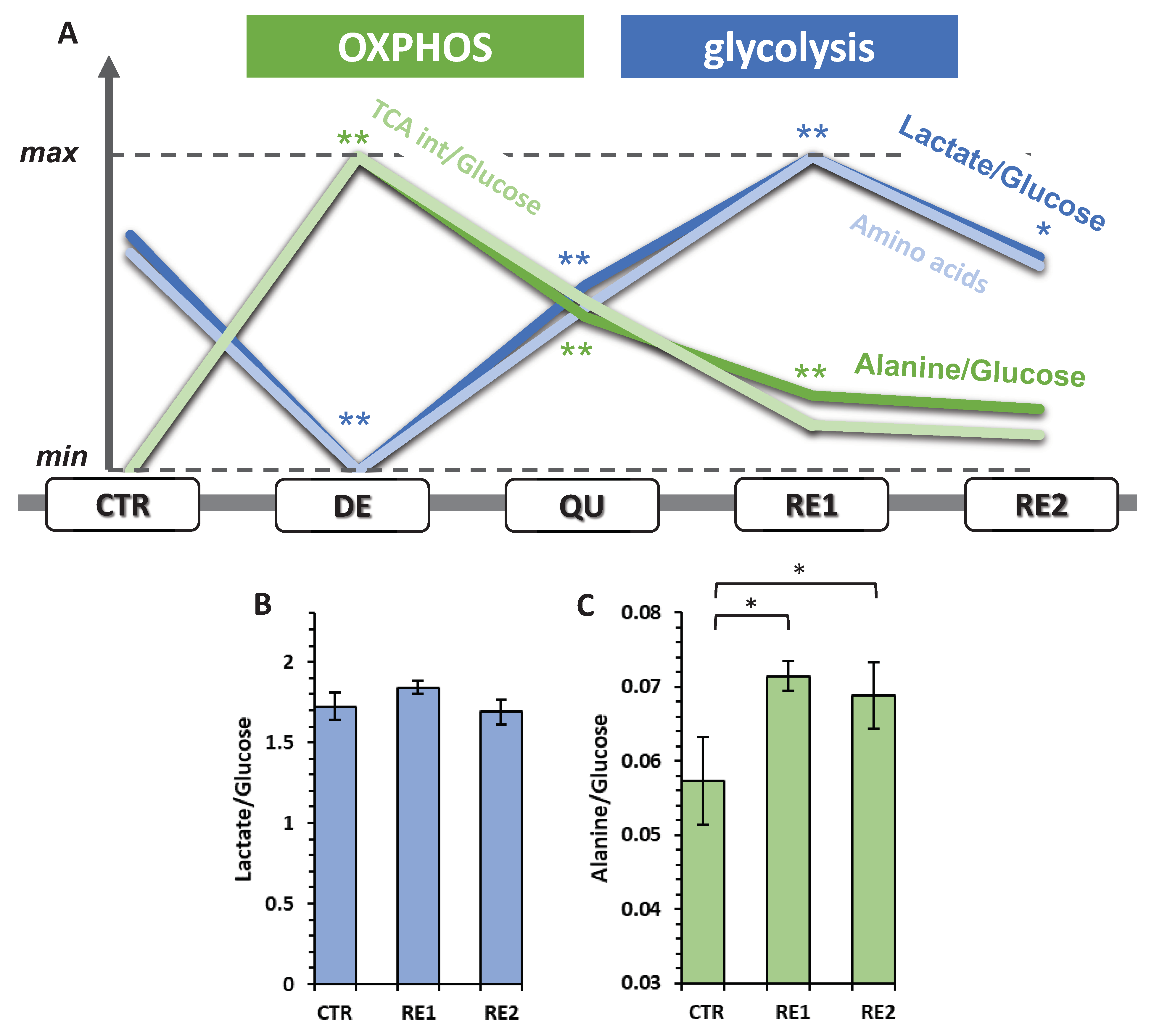
3.6. The Recurrent Glycolysis/OXPHOS Switch of Cells after Etoposide Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 2021. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, C.-Y.; Yuan, F. Mathematical Modeling of the Phoenix Rising Pathway. PLoS Comput. Biol. 2014, 10, 1003461. [Google Scholar] [CrossRef] [PubMed]
- Berish, R.B.; Ali, A.N.; Telmer, P.G.; Ronald, J.A.; Leong, H.S. Translational models of prostate cancer bone metastasis. Nat. Rev. Urol. 2018, 15, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem.—Anti-Cancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef]
- Kamradt, J.M.; Pienta, K.J. Etoposide in prostate cancer. Expert Opin. Pharmacother. 2005, 1, 271–275. [Google Scholar] [CrossRef]
- Cattrini, C.; Capaia, M.; Boccardo, F.; Barboro, P. Etoposide and topoisomerase II inhibition for aggressive prostate cancer: Data from a translational study. Cancer Treat. Res. Commun. 2020, 25, 100221. [Google Scholar] [CrossRef]
- Corsi, F.; Capradossi, F.; Pelliccia, A.; Briganti, S.; Bruni, E.; Traversa, E.; Torino, F.; Reichle, A.; Ghibelli, L. Apoptosis as a Driver of Therapy-Induced Cancer Repopulation and Acquired Cell-Resistance (CRAC): A Simple In Vitro Model of Phoenix Rising in Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 1152. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368. [Google Scholar] [CrossRef]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef]
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. Heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef]
- Petrella, G.; Ciufolini, G.; Vago, R.; Cicero, D.O. The Interplay between Oxidative Phosphorylation and Glycolysis as a Potential Marker of Bladder Cancer Progression. Int. J. Mol. Sci. 2020, 21, 8107. [Google Scholar] [CrossRef] [PubMed]
- Kdadra, M.; Höckner, S.; Leung, H.; Kremer, W.; Schiffer, E. Metabolomics biomarkers of prostate cancer: A systematic review. Diagnostics 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Gupta, A.; Mandhani, A.; Sankhwar, S.N. Metabolomics-derived prostate cancer biomarkers: Fact or fiction? J. Proteome Res. 2015, 14, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef]
- Dolfi, S.C.; Li, L.; Chan, -Y.; Qiu, J.; Tedeschi, P.M.; Bertino, J.R.; Hirshfield, K.M.; Oltvai, Z.N.; Vazquez, A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013, 1, 1–13. [Google Scholar] [CrossRef]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays 2013, 35, 1050–1055. [Google Scholar] [CrossRef]
- Strmiska, V.; Michalek, P.; Eckschlager, T.; Stiborova, M.; Adam, V.; Krizkova, S.; Heger, Z. Prostate cancer-specific hallmarks of amino acids metabolism: Towards a paradigm of precision medicine. Biochim. Biophys. Acta—Rev. Cancer 2019, 1871, 248–258. [Google Scholar] [CrossRef]
- Yadav, N.; Kumar, S.; Marlowe, T.; Chaudhary, A.K.; Kumar, R.; Wang, J.; O’Malley, J.; Boland, P.M.; Jayanthi, S.; Kumar, T.K.S.; et al. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015, 6, e1969. [Google Scholar] [CrossRef]
- Spragg, R.G.; Hinshaw, D.B.; Hyslop, P.A.; Schraufstätter, I.U.; Cochrane, C.G. Alterations in adenosine triphosphate and energy charge in cultured endothelial and P388D1 cells after oxidant injury. J. Clin. Investig. 1985, 76, 1471–1476. [Google Scholar] [CrossRef]
- SCHUPPE-KOISTINEN, I.; MOLDEUS, P.; BERGMAN, T.; COTGREAVE, I.A. S-Thiolation of human endothelial cell glyceraldehyde-3-phosphate dehydrogenase after hydrogen peroxide treatment. Eur. J. Biochem. 1994, 221, 1033–1037. [Google Scholar] [CrossRef]
- Colussi, C.; Albertini, M.C.; Coppola, S.; Rovidati, S.; Galli, F.; Ghibelli, L. H2O2-induced block of glycolysis as an active ADP-ribosylation reaction protecting cells from apoptosis. FASEB J. 2000, 14, 2266–2276. [Google Scholar] [CrossRef]
- Pradelli, L.A.; Villa, E.; Zunino, B.; Marchetti, S.; Ricci, J.E. Glucose metabolism is inhibited by caspases upon the induction of apoptosis. Cell Death Dis. 2014, 5, e1406-7. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Li, Q.; Wu, F.; Lin, J.; Chen, J.; Zheng, H.; Guo, L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Scimè, A. Metabolic Regulation of Epithelial to Mesenchymal Transition: Implications for Endocrine Cancer. Front. Endocrinol. (Lausanne) 2019, 10. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Jeong, E.K.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S.; Lee, S.Y.; Jeon, H.M.; et al. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget 2016, 7, 7925–7939. [Google Scholar] [CrossRef]
- Neinast, M.; Murashige, D.; Arany, Z. Branched Chain Amino Acids. Annu. Rev. Physiol. 2019, 81, 139–164. [Google Scholar] [CrossRef]
- Fox, D.B.; Alvarez, J.V. Epithelial-to-mesenchymal transition activates Bcat1 expression to promote recurrent tumor growth. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sun, H.; Yang, X.; Liang, L.; Zhang, M.; Li, Y.; Chen, J.; Wang, F.; Yang, T.; Meng, F.; Lai, X.; et al. Metabolic switch and epithelial–mesenchymal transition cooperate to regulate pluripotency. EMBO J. 2020, 39. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Cha, Y.H.; Lee, J.; Lee, S.H.; Yang, J.H.; Yun, J.S.; Cho, E.S.; Zhang, X.; Nam, M.; Kim, N.; et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gaude, E.; Frezza, C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aspuria, P.-J.P.; Lunt, S.Y.; Väremo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 1–15. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; Da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Jones, W.; Bianchi, K. Aerobic Glycolysis: Beyond Proliferation. Front. Immunol. 2015, 6, 1–5. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Huo, H.Y.; Ao, S.; Liu, T.; Yang, L.; Fei, Z.Y.; Zhang, Z.Q.; Ding, L.; Cui, Q.H.; Lin, J.; et al. TGF-β1-induced epithelial-mesenchymal transition increases fatty acid oxidation and OXPHOS activity via the p-AMPK pathway in breast cancer cells. Oncol. Rep. 2020, 44, 1206–1215. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrella, G.; Corsi, F.; Ciufolini, G.; Germini, S.; Capradossi, F.; Pelliccia, A.; Torino, F.; Ghibelli, L.; Cicero, D.O. Metabolic Reprogramming of Castration-Resistant Prostate Cancer Cells as a Response to Chemotherapy. Metabolites 2023, 13, 65. https://doi.org/10.3390/metabo13010065
Petrella G, Corsi F, Ciufolini G, Germini S, Capradossi F, Pelliccia A, Torino F, Ghibelli L, Cicero DO. Metabolic Reprogramming of Castration-Resistant Prostate Cancer Cells as a Response to Chemotherapy. Metabolites. 2023; 13(1):65. https://doi.org/10.3390/metabo13010065
Chicago/Turabian StylePetrella, Greta, Francesca Corsi, Giorgia Ciufolini, Sveva Germini, Francesco Capradossi, Andrea Pelliccia, Francesco Torino, Lina Ghibelli, and Daniel Oscar Cicero. 2023. "Metabolic Reprogramming of Castration-Resistant Prostate Cancer Cells as a Response to Chemotherapy" Metabolites 13, no. 1: 65. https://doi.org/10.3390/metabo13010065
APA StylePetrella, G., Corsi, F., Ciufolini, G., Germini, S., Capradossi, F., Pelliccia, A., Torino, F., Ghibelli, L., & Cicero, D. O. (2023). Metabolic Reprogramming of Castration-Resistant Prostate Cancer Cells as a Response to Chemotherapy. Metabolites, 13(1), 65. https://doi.org/10.3390/metabo13010065