
Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders



Abstract
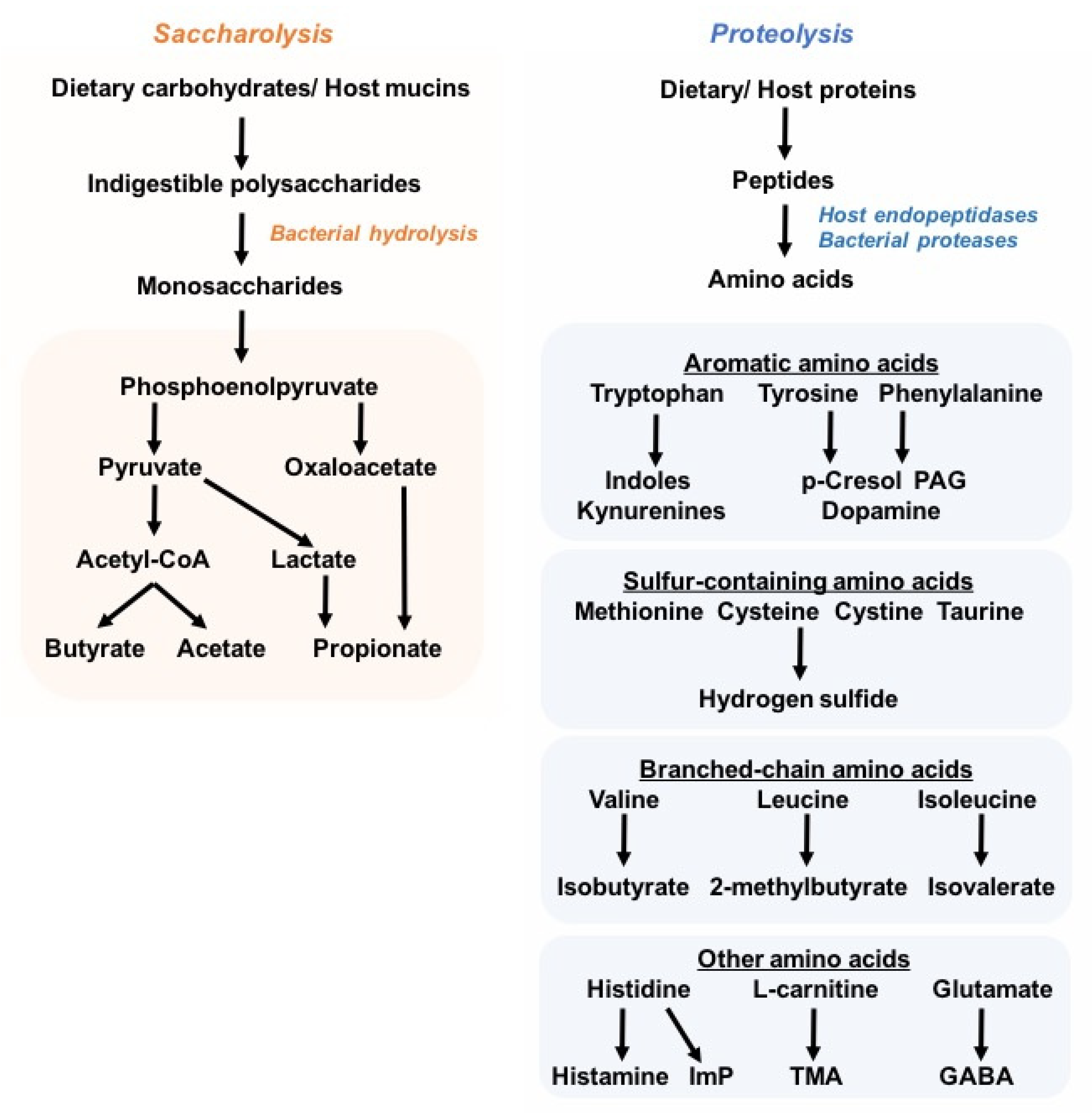
:1. Introduction
2. Analytical Approaches Aiding Microbiota–Metabolome Studies
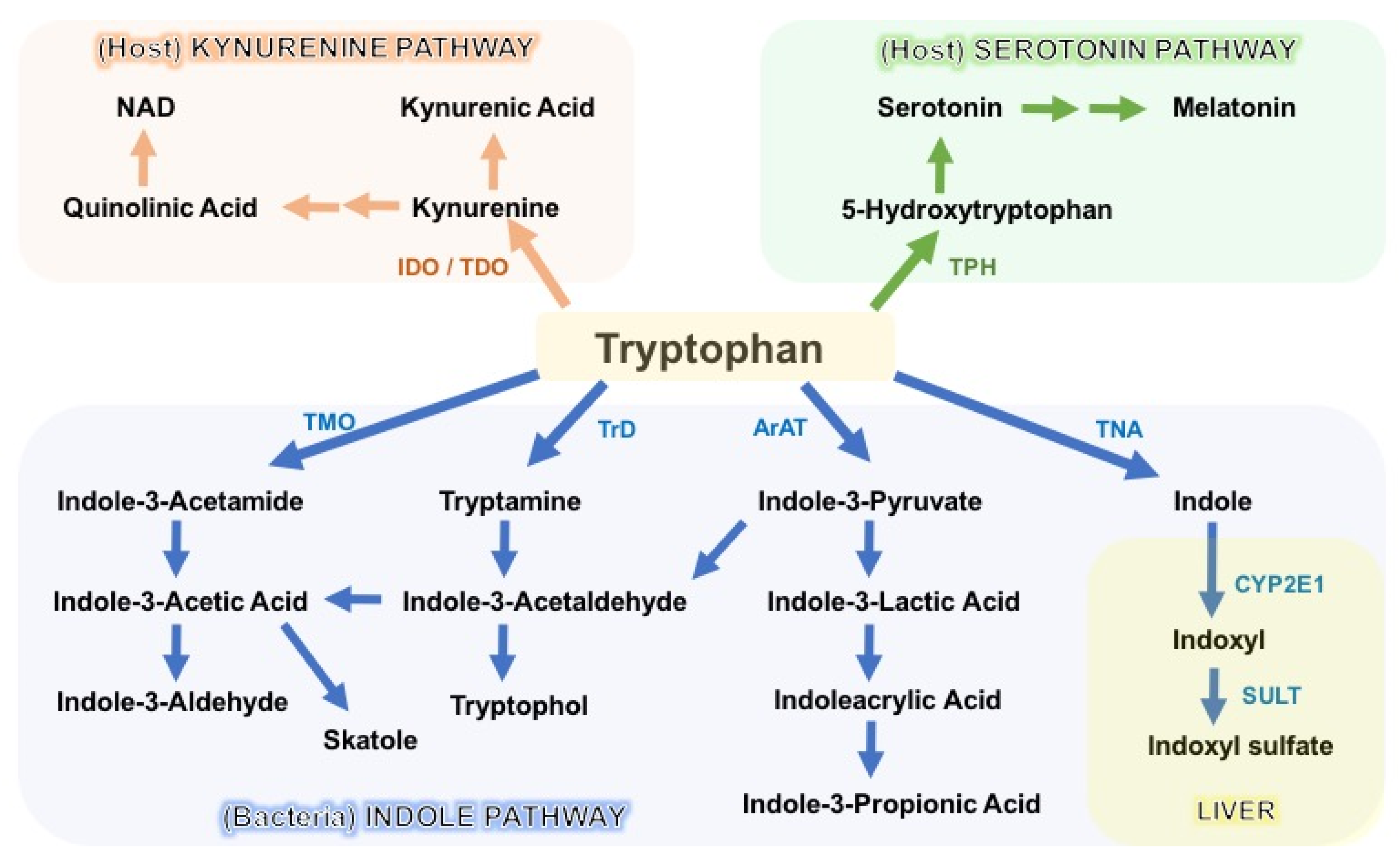
3. Tryptophan Catabolism
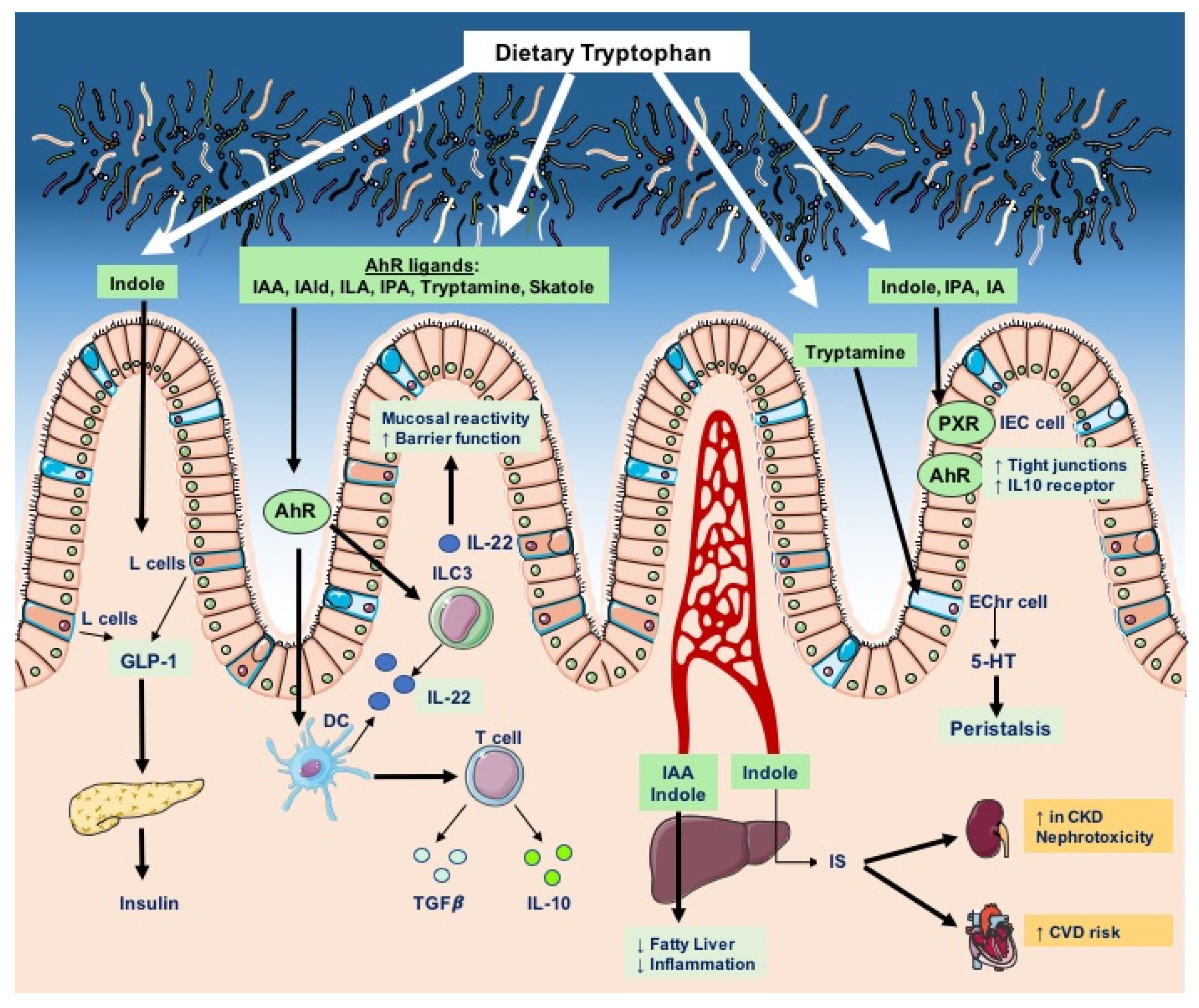
4. Tryptophan Catabolites as AhR Ligands: Role in Intestinal Homeostasis
5. Tryptophan Catabolites in Intestinal Homeostasis and Inflammation
6. Tryptophan Catabolites in Metabolic Disorders
7. Tryptophan Catabolites in Chronic Kidney Diseases and Cardiovascular Diseases
8. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Tierney, B.T.; Yang, Z.; Luber, J.M.; Beaudin, M.; Wibowo, M.C.; Baek, C.; Mehlenbacher, E.; Patel, C.J.; Kostic, A.D. The Landscape of Genetic Content in the Gut and Oral Human Microbiome. Cell Host Microbe 2019, 26, 283–295.e288. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef] [PubMed]
- Fenneman, A.C.; Rampanelli, E.; Yin, Y.S.; Ames, J.; Blaser, M.J.; Fliers, E.; Nieuwdorp, M. Gut microbiota and metabolites in the pathogenesis of endocrine disease. Biochem. Soc. Trans. 2020, 48, 915–931. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Mosterd, C.M.; Kanbay, M.; van den Born, B.J.H.; van Raalte, D.H.; Rampanelli, E. Intestinal microbiota and diabetic kidney diseases: The Role of microbiota and derived metabolites inmodulation of renal inflammation and disease progression. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101484. [Google Scholar] [CrossRef]
- He, Y.; Wu, W.; Zheng, H.M.; Li, P.; McDonald, D.; Sheng, H.F.; Chen, M.X.; Chen, Z.H.; Ji, G.Y.; Zheng, Z.D.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef]
- Deschasaux, M.; Bouter, K.E.; Prodan, A.; Levin, E.; Groen, A.K.; Herrema, H.; Tremaroli, V.; Bakker, G.J.; Attaye, I.; Pinto-Sietsma, S.J.; et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 2018, 24, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Kim, J.; Duffy, P.R.; Schlegel, V.L.; Walter, J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS ONE 2010, 5, e15046. [Google Scholar] [CrossRef] [Green Version]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e910. [Google Scholar] [CrossRef] [PubMed]
- Deehan, E.C.; Yang, C.; Perez-Munoz, M.E.; Nguyen, N.K.; Cheng, C.C.; Triador, L.; Zhang, Z.; Bakal, J.A.; Walter, J. Precision Microbiome Modulation with Discrete Dietary Fiber Structures Directs Short-Chain Fatty Acid Production. Cell Host Microbe 2020, 27, 389–404.e386. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, T.L.; Kunstner, A.; Kennedy, J.J.; Fang, Q.; Asarian, L.; Culp-Hill, R.; D’Alessandro, A.; Teuscher, C.; Busch, H.; Krementsov, D.N. Interactions between host genetics and gut microbiota determine susceptibility to CNS autoimmunity. Proc. Natl. Acad. Sci. USA 2020, 117, 27516–27527. [Google Scholar] [CrossRef]
- Wilmanski, T.; Diener, C.; Rappaport, N.; Patwardhan, S.; Wiedrick, J.; Lapidus, J.; Earls, J.C.; Zimmer, A.; Glusman, G.; Robinson, M.; et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metab. 2021, 3, 274–286. [Google Scholar] [CrossRef]
- Rajilic-Stojanovic, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef]
- Molinaro, A.; Bel Lassen, P.; Henricsson, M.; Wu, H.; Adriouch, S.; Belda, E.; Chakaroun, R.; Nielsen, T.; Bergh, P.O.; Rouault, C.; et al. Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat. Commun. 2020, 11, 5881. [Google Scholar] [CrossRef]
- Menni, C.; Zhu, J.; Le Roy, C.I.; Mompeo, O.; Young, K.; Rebholz, C.M.; Selvin, E.; North, K.E.; Mohney, R.P.; Bell, J.T.; et al. Serum metabolites reflecting gut microbiome alpha diversity predict type 2 diabetes. Gut Microbes 2020, 11, 1632–1642. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Deleu, S.; Machiels, K.; Raes, J.; Verbeke, K.; Vermeire, S. Short chain fatty acids and its producing organisms: An overlooked therapy for IBD? EBioMedicine 2021, 66, 103293. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Randrianarisoa, E.; Lehn-Stefan, A.; Wang, X.; Hoene, M.; Peter, A.; Heinzmann, S.S.; Zhao, X.; Konigsrainer, I.; Konigsrainer, A.; Balletshofer, B.; et al. Relationship of Serum Trimethylamine N-Oxide (TMAO) Levels with early Atherosclerosis in Humans. Sci. Rep. 2016, 6, 26745. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Stahlman, M.; Khan, M.T.; Schmidt, C.; Manneras-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e917. [Google Scholar] [CrossRef]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e822. [Google Scholar] [CrossRef]
- Poesen, R.; Claes, K.; Evenepoel, P.; de Loor, H.; Augustijns, P.; Kuypers, D.; Meijers, B. Microbiota-Derived Phenylacetylglutamine Associates with Overall Mortality and Cardiovascular Disease in Patients with CKD. J. Am. Soc. Nephrol. 2016, 27, 3479–3487. [Google Scholar] [CrossRef]
- Ottosson, F.; Brunkwall, L.; Smith, E.; Orho-Melander, M.; Nilsson, P.M.; Fernandez, C.; Melander, O. The gut microbiota-related metabolite phenylacetylglutamine associates with increased risk of incident coronary artery disease. J. Hypertens. 2020, 38, 2427–2434. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Lee, C.C.; Sun, C.Y.; Hsu, H.J.; Tsai, C.J.; Tzen, C.Y.; Wang, Y.C.; Lin, C.Y.; Wu, M.S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Aronov, P.A.; Luo, F.J.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Mare, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- Bui, T.P.; Ritari, J.; Boeren, S.; de Waard, P.; Plugge, C.M.; de Vos, W.M. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat. Commun. 2015, 6, 10062. [Google Scholar] [CrossRef]
- Milshteyn, A.; Colosimo, D.A.; Brady, S.F. Accessing Bioactive Natural Products from the Human Microbiome. Cell Host Microbe 2018, 23, 725–736. [Google Scholar] [CrossRef]
- Fujisaka, S.; Avila-Pacheco, J.; Soto, M.; Kostic, A.; Dreyfuss, J.M.; Pan, H.; Ussar, S.; Altindis, E.; Li, N.; Bry, L.; et al. Diet, Genetics, and the Gut Microbiome Drive Dynamic Changes in Plasma Metabolites. Cell Rep. 2018, 22, 3072–3086. [Google Scholar] [CrossRef]
- Zierer, J.; Jackson, M.A.; Kastenmuller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.I.; Weir, W.H.; Crowley, J.R.; Hink, T.; Reske, K.A.; Kwon, J.H.; Burnham, C.D.; Dubberke, E.R.; Mucha, P.J.; Henderson, J.P. Metabolomic networks connect host-microbiome processes to human Clostridioides difficile infections. J. Clin. Investig. 2019, 129, 3792–3806. [Google Scholar] [CrossRef]
- Zhou, B.; Xiao, J.F.; Tuli, L.; Ressom, H.W. LC-MS-based metabolomics. Mol. Biosyst. 2012, 8, 470–481. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30.4.1–30.4.2. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Q.; Hsu, C.C. Can Diet Influence Our Health by Altering Intestinal Microbiota-Derived Fecal Metabolites? mSystems 2018, 3, e00187-17. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Claus, D.; Geypens, B.; Hiele, M.; Geboes, K.; Rutgeerts, P.; Ghoos, Y. Amount and fate of egg protein escaping assimilation in the small intestine of humans. Am. J. Physiol. 1999, 277, G935–G943. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.A.; Sladen, G.E.; Dawson, A.M. Protein absorption and ammonia production: The effects of dietary protein and removal of the colon. Br. J. Nutr. 1976, 35, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Macfarlane, G.T. The control and consequences of bacterial fermentation in the human colon. J. Appl. Bacteriol. 1991, 70, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Diether, N.E.; Willing, B.P. Microbial Fermentation of Dietary Protein: An Important Factor in Diet-Microbe-Host Interaction. Microorganisms 2019, 7, 19. [Google Scholar] [CrossRef]
- Roager, H.M.; Hansen, L.B.; Bahl, M.I.; Frandsen, H.L.; Carvalho, V.; Gobel, R.J.; Dalgaard, M.D.; Plichta, D.R.; Sparholt, M.H.; Vestergaard, H.; et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat. Microbiol. 2016, 1, 16093. [Google Scholar] [CrossRef]
- Riva, A.; Kuzyk, O.; Forsberg, E.; Siuzdak, G.; Pfann, C.; Herbold, C.; Daims, H.; Loy, A.; Warth, B.; Berry, D. A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome. Nat. Commun. 2019, 10, 4366. [Google Scholar] [CrossRef]
- Geypens, B.; Claus, D.; Evenepoel, P.; Hiele, M.; Maes, B.; Peeters, M.; Rutgeerts, P.; Ghoos, Y. Influence of dietary protein supplements on the formation of bacterial metabolites in the colon. Gut 1997, 41, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef] [PubMed]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e1321. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.H.W.; et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Saigusa, D.; Kanemitsu, Y.; Matsumoto, Y.; Thanai, P.; Suzuki, N.; Mise, K.; Yamaguchi, H.; Nakamura, T.; Asaji, K.; et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Miikeda, A.; Zuckerman, J.; Jia, X.; Charugundla, S.; Zhou, Z.; Kaczor-Urbanowicz, K.E.; Magyar, C.; Guo, F.; Wang, Z.; et al. Inhibition of microbiota-dependent TMAO production attenuates chronic kidney disease in mice. Sci. Rep. 2021, 11, 518. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Smith, T. A Modification of the Method for Determining the Production of Indol by Bacteria. J. Exp. Med. 1897, 2, 543–547. [Google Scholar] [CrossRef]
- Russell, W.R.; Duncan, S.H.; Scobbie, L.; Duncan, G.; Cantlay, L.; Calder, A.G.; Anderson, S.E.; Flint, H.J. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol. Nutr. Food Res. 2013, 57, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Elsden, S.R.; Hilton, M.G.; Waller, J.M. The end products of the metabolism of aromatic amino acids by Clostridia. Arch. Microbiol. 1976, 107, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Aragozzini, F.; Ferrari, A.; Pacini, N.; Gualandris, R. Indole-3-lactic acid as a tryptophan metabolite produced by Bifidobacterium spp. Appl. Environ. Microbiol. 1979, 38, 544–546. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8alphaalpha(+) T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, T.R.; Price, N.P.; Drake, H.L.; Cotta, M.A. Catabolic pathway for the production of skatole and indoleacetic acid by the acetogen Clostridium drakei, Clostridium scatologenes, and swine manure. Appl. Environ. Microbiol. 2008, 74, 1950–1953. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Vujkovic-Cvijin, I.; Sortino, O.; Verheij, E.; Sklar, J.; Wit, F.W.; Kootstra, N.A.; Sellers, B.; Brenchley, J.M.; Ananworanich, J.; Loeff, M.S.V.; et al. HIV-associated gut dysbiosis is independent of sexual practice and correlates with noncommunicable diseases. Nat. Commun. 2020, 11, 2448. [Google Scholar] [CrossRef]
- Metghalchi, S.; Ponnuswamy, P.; Simon, T.; Haddad, Y.; Laurans, L.; Clement, M.; Dalloz, M.; Romain, M.; Esposito, B.; Koropoulis, V.; et al. Indoleamine 2,3-Dioxygenase Fine-Tunes Immune Homeostasis in Atherosclerosis and Colitis through Repression of Interleukin-10 Production. Cell Metab 2015, 22, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.T.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab. 2018, 27, 378–392.e375. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Femenia, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. The footprint of kynurenine pathway in every cancer: A new target for chemotherapy. Eur. J. Pharmacol. 2021, 896, 173921. [Google Scholar] [CrossRef] [PubMed]
- Hornigold, N.; Dunn, K.R.; Craven, R.A.; Zougman, A.; Trainor, S.; Shreeve, R.; Brown, J.; Sewell, H.; Shires, M.; Knowles, M.; et al. Dysregulation at multiple points of the kynurenine pathway is a ubiquitous feature of renal cancer: Implications for tumour immune evasion. Br. J. Cancer 2020, 123, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Favennec, M.; Hennart, B.; Caiazzo, R.; Leloire, A.; Yengo, L.; Verbanck, M.; Arredouani, A.; Marre, M.; Pigeyre, M.; Bessede, A.; et al. The kynurenine pathway is activated in human obesity and shifted toward kynurenine monooxygenase activation. Obesity 2015, 23, 2066–2074, Erratum in Obesity 2016, 24, 1821. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.E.; Astola, N.; Cribbs, A.P.; Goddard, M.E.; Park, I.; Green, P.; Davies, A.H.; Williams, R.O.; Feldmann, M.; Monaco, C. Indoleamine 2,3-dioxygenase-1 is protective in atherosclerosis and its metabolites provide new opportunities for drug development. Proc. Natl. Acad. Sci. USA 2015, 112, 13033–13038. [Google Scholar] [CrossRef] [PubMed]
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut--functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.A.; Nguyen, J.C.; Polglaze, K.E.; Bertrand, P.P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Sjogren, K.; Engdahl, C.; Henning, P.; Lerner, U.H.; Tremaroli, V.; Lagerquist, M.K.; Backhed, F.; Ohlsson, C. The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 2012, 27, 1357–1367. [Google Scholar] [CrossRef] [Green Version]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef]
- Takaki, M.; Mawe, G.M.; Barasch, J.M.; Gershon, M.D.; Gershon, M.D. Physiological responses of guinea-pig myenteric neurons secondary to the release of endogenous serotonin by tryptamine. Neuroscience 1985, 16, 223–240. [Google Scholar] [CrossRef]
- Devlin, A.S.; Marcobal, A.; Dodd, D.; Nayfach, S.; Plummer, N.; Meyer, T.; Pollard, K.S.; Sonnenburg, J.L.; Fischbach, M.A. Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota. Cell Host Microbe 2016, 20, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Luo, C.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e26. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Backhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Darkoh, C.; Chappell, C.; Gonzales, C.; Okhuysen, P. A rapid and specific method for the detection of indole in complex biological samples. Appl. Environ. Microbiol. 2015, 81, 8093–8097. [Google Scholar] [CrossRef]
- Rosas, H.D.; Doros, G.; Bhasin, S.; Thomas, B.; Gevorkian, S.; Malarick, K.; Matson, W.; Hersch, S.M. A systems-level “misunderstanding”: The plasma metabolome in Huntington’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 756–768. [Google Scholar] [CrossRef]
- Pavlova, T.; Vidova, V.; Bienertova-Vasku, J.; Janku, P.; Almasi, M.; Klanova, J.; Spacil, Z. Urinary intermediates of tryptophan as indicators of the gut microbial metabolism. Anal. Chim. Acta 2017, 987, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Anesi, A.; Rubert, J.; Oluwagbemigun, K.; Orozco-Ruiz, X.; Nothlings, U.; Breteler, M.M.B.; Mattivi, F. Metabolic Profiling of Human Plasma and Urine, Targeting Tryptophan, Tyrosine and Branched Chain Amino Acid Pathways. Metabolites 2019, 9, 261. [Google Scholar] [CrossRef]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef] [PubMed]
- Jennis, M.; Cavanaugh, C.R.; Leo, G.C.; Mabus, J.R.; Lenhard, J.; Hornby, P.J. Microbiota-derived tryptophan indoles increase after gastric bypass surgery and reduce intestinal permeability in vitro and in vivo. Neurogastroenterol. Motil. 2018, 30, e13178. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, T.K.; Rao, G.R. Beta-indoleethanol and beta-indolelactic acid production by Candida species: Their antibacterial and autoantibiotic action. Antimicrob. Agents Chemother. 1976, 9, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Honore, A.H.; Aunsbjerg, S.D.; Ebrahimi, P.; Thorsen, M.; Benfeldt, C.; Knochel, S.; Skov, T. Metabolic footprinting for investigation of antifungal properties of Lactobacillus paracasei. Anal. Bioanal. Chem. 2016, 408, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Xu, C.; Zhang, X. The effect of tryptophol on the bacteriophage infection in high-temperature environment. Appl. Microbiol. Biotechnol. 2015, 99, 8101–8111. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-kappaB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- McBerry, C.; Gonzalez, R.M.; Shryock, N.; Dias, A.; Aliberti, J. SOCS2-induced proteasome-dependent TRAF6 degradation: A common anti-inflammatory pathway for control of innate immune responses. PLoS ONE 2012, 7, e38384. [Google Scholar] [CrossRef]
- Kimura, A.; Naka, T.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9721–9726. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, H.; Fan, H.; Zuo, D.; Shou, Z.; Liao, Y.; Nan, Z.; Tang, Q. The role of STAT3 and AhR in the differentiation of CD4+ T cells into Th17 and Treg cells. Medicine 2017, 96, e6615. [Google Scholar] [CrossRef] [PubMed]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, D.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat. Med. 2015, 21, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362.e355. [Google Scholar] [CrossRef]
- Chng, S.H.; Kundu, P.; Dominguez-Brauer, C.; Teo, W.L.; Kawajiri, K.; Fujii-Kuriyama, Y.; Mak, T.W.; Pettersson, S. Ablating the aryl hydrocarbon receptor (AhR) in CD11c+ cells perturbs intestinal epithelium development and intestinal immunity. Sci. Rep. 2016, 6, 23820. [Google Scholar] [CrossRef]
- Lanis, J.M.; Alexeev, E.E.; Curtis, V.F.; Kitzenberg, D.A.; Kao, D.J.; Battista, K.D.; Gerich, M.E.; Glover, L.E.; Kominsky, D.J.; Colgan, S.P. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 2017, 10, 1133–1144. [Google Scholar] [CrossRef]
- Sujino, T.; London, M.; Hoytema van Konijnenburg, D.P.; Rendon, T.; Buch, T.; Silva, H.M.; Lafaille, J.J.; Reis, B.S.; Mucida, D. Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science 2016, 352, 1581–1586. [Google Scholar] [CrossRef]
- Chen, W.; Pu, A.; Sheng, B.; Zhang, Z.; Li, L.; Liu, Z.; Wang, Q.; Li, X.; Ma, Y.; Yu, M.; et al. Aryl hydrocarbon receptor activation modulates CD8alphaalpha(+)TCRalphabeta(+) IELs and suppression of colitis manifestations in mice. Biomed. Pharmacother. 2017, 87, 127–134. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011, 13, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Pang, Z.; Shu, W.; Wu, W.; Sun, M.; Cong, Y.; Liu, Z. Anti-TNF Therapy Induces CD4+ T-Cell Production of IL-22 and Promotes Epithelial Repairs in Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248.e231. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Wagage, S.; Harms Pritchard, G.; Dawson, L.; Buza, E.L.; Sonnenberg, G.F.; Hunter, C.A. The Group 3 Innate Lymphoid Cell Defect in Aryl Hydrocarbon Receptor Deficient Mice Is Associated with T Cell Hyperactivation during Intestinal Infection. PLoS ONE 2015, 10, e0128335. [Google Scholar] [CrossRef]
- Qiu, J.; Guo, X.; Chen, Z.M.; He, L.; Sonnenberg, G.F.; Artis, D.; Fu, Y.X.; Zhou, L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 2013, 39, 386–399. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Li, J.; Doty, A.; Glover, S.C. Aryl hydrocarbon receptor signaling involves in the human intestinal ILC3/ILC1 conversion in the inflamed terminal ileum of Crohn’s disease patients. Inflamm. Cell Signal. 2016, 3. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.K.; Thaker, A.I.; Kanuri, N.; Riehl, T.E.; Rowley, C.W.; Stenson, W.F.; Ciorba, M.A. Serum analysis of tryptophan catabolism pathway: Correlation with Crohn’s disease activity. Inflamm. Bowel Dis. 2012, 18, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Hasler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e1502. [Google Scholar] [CrossRef] [PubMed]
- Brawner, K.M.; Yeramilli, V.A.; Duck, L.W.; Van Der Pol, W.; Smythies, L.E.; Morrow, C.D.; Elson, C.O.; Martin, C.A. Depletion of dietary aryl hydrocarbon receptor ligands alters microbiota composition and function. Sci. Rep. 2019, 9, 14724. [Google Scholar] [CrossRef] [PubMed]
- Culbreath, C.; Tanner, S.M.; Yeramilli, V.A.; Berryhill, T.F.; Lorenz, R.G.; Martin, C.A. Environmental-mediated intestinal homeostasis in neonatal mice. J. Surg. Res. 2015, 198, 494–501. [Google Scholar] [CrossRef]
- Takamura, T.; Harama, D.; Fukumoto, S.; Nakamura, Y.; Shimokawa, N.; Ishimaru, K.; Ikegami, S.; Makino, S.; Kitamura, M.; Nakao, A. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol. Cell Biol. 2011, 89, 817–822. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Powell, D.N.; Swimm, A.; Sonowal, R.; Bretin, A.; Gewirtz, A.T.; Jones, R.M.; Kalman, D. Indoles from the commensal microbiota act via the AHR and IL-10 to tune the cellular composition of the colonic epithelium during aging. Proc. Natl. Acad. Sci. USA 2020, 117, 21519–21526. [Google Scholar] [CrossRef]
- Xiao, H.W.; Cui, M.; Li, Y.; Dong, J.L.; Zhang, S.Q.; Zhu, C.C.; Jiang, M.; Zhu, T.; Wang, B.; Wang, H.C.; et al. Gut microbiota-derived indole 3-propionic acid protects against radiation toxicity via retaining acyl-CoA-binding protein. Microbiome 2020, 8, 69. [Google Scholar] [CrossRef]
- Guo, H.; Chou, W.C.; Lai, Y.; Liang, K.; Tam, J.W.; Brickey, W.J.; Chen, L.; Montgomery, N.D.; Li, X.; Bohannon, L.M.; et al. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science 2020, 370, eaay9097. [Google Scholar] [CrossRef]
- Jin, U.H.; Lee, S.O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol. 2014, 85, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Gensollen, T.; Gandhi, A.; Oh, S.F.; Neves, J.F.; Collin, F.; Lavin, R.; Serra, C.; Glickman, J.; de Silva, P.S.A.; et al. Dietary and Microbial Oxazoles Induce Intestinal Inflammation by Modulating Aryl Hydrocarbon Receptor Responses. Cell 2018, 173, 1123–1134.e1111. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Hwang, Y.J.; Yun, M.O.; Kim, J.H.; Oh, G.S.; Park, J.H. Indoxyl 3-sulfate stimulates Th17 differentiation enhancing phosphorylation of c-Src and STAT3 to worsen experimental autoimmune encephalomyelitis. Toxicol. Lett. 2013, 220, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e917. [Google Scholar] [CrossRef] [PubMed]
- Kootte, R.S.; Levin, E.; Salojarvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619.e616. [Google Scholar] [CrossRef]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Tremaroli, V.; Schmidt, C.; Lundqvist, A.; Olsson, L.M.; Kramer, M.; Gummesson, A.; Perkins, R.; Bergstrom, G.; Backhed, F. The Gut Microbiota in Prediabetes and Diabetes: A Population-Based Cross-Sectional Study. Cell Metab. 2020, 32, 379–390.e373. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef]
- den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch from Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Ducastel, S.; Touche, V.; Trabelsi, M.S.; Boulinguiez, A.; Butruille, L.; Nawrot, M.; Peschard, S.; Chavez-Talavera, O.; Dorchies, E.; Vallez, E.; et al. The nuclear receptor FXR inhibits Glucagon-Like Peptide-1 secretion in response to microbiota-derived Short-Chain Fatty Acids. Sci. Rep. 2020, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef]
- Mallmann, N.H.; Lima, E.S.; Lalwani, P. Dysregulation of Tryptophan Catabolism in Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2018, 16, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Laurans, L.; Venteclef, N.; Haddad, Y.; Chajadine, M.; Alzaid, F.; Metghalchi, S.; Sovran, B.; Denis, R.G.P.; Dairou, J.; Cardellini, M.; et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat. Med. 2018, 24, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.; Agus, A.; Planchais, J.; Lamas, B.; Jarry, A.C.; Martin, R.; Michel, M.L.; Chong-Nguyen, C.; Roussel, R.; Straube, M.; et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab. 2018, 28, 737–749.e734. [Google Scholar] [CrossRef] [PubMed]
- de Mello, V.D.; Paananen, J.; Lindstrom, J.; Lankinen, M.A.; Shi, L.; Kuusisto, J.; Pihlajamaki, J.; Auriola, S.; Lehtonen, M.; Rolandsson, O.; et al. Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci. Rep. 2017, 7, 46337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, M.; Tanaka, M.; Toda, H.; Asano, M.; Yamazaki, M.; Hasegawa, G.; Imai, S.; Nakamura, N. High plasma 5-hydroxyindole-3-acetic acid concentrations in subjects with metabolic syndrome. Diabetes Care 2012, 35, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Wu, V.; Wu, P.C.; Wu, C.J. Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.S.; Chen, J.; Zou, J.Z.; Zhong, Y.H.; Teng, J.; Ji, J.; Chen, Z.W.; Liu, Z.H.; Shen, B.; Nie, Y.X.; et al. Association of indoxyl sulfate with heart failure among patients on hemodialysis. Clin. J. Am. Soc. Nephrol. 2015, 10, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work, G. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Virtue, A.T.; McCright, S.J.; Wright, J.M.; Jimenez, M.T.; Mowel, W.K.; Kotzin, J.J.; Joannas, L.; Basavappa, M.G.; Spencer, S.P.; Clark, M.L.; et al. The gut microbiota regulates white adipose tissue inflammation and obesity via a family of microRNAs. Sci. Transl. Med. 2019, 11, eaav1892. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, A.; Elfving, B.; Hokland, M.; Wegener, G.; Lund, S. The microbial metabolite indole-3-propionic acid improves glucose metabolism in rats, but does not affect behaviour. Arch. Physiol. Biochem. 2018, 124, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Gribble, F.M.; Reimann, F. Metabolic Messengers: Glucagon-like peptide 1. Nat. Metab. 2021, 3, 142–148. [Google Scholar] [CrossRef]
- Postal, B.G.; Ghezzal, S.; Aguanno, D.; Andre, S.; Garbin, K.; Genser, L.; Brot-Laroche, E.; Poitou, C.; Soula, H.; Leturque, A.; et al. AhR activation defends gut barrier integrity against damage occurring in obesity. Mol. Metab. 2020, 39, 101007. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ota, N.; Manzanillo, P.; Kates, L.; Zavala-Solorio, J.; Eidenschenk, C.; Zhang, J.; Lesch, J.; Lee, W.P.; Ross, J.; et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014, 514, 237–241. [Google Scholar] [CrossRef]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.M.; Reimann, F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, C.; Neyrinck, A.M.; Leyrolle, Q.; Baldin, P.; Leclercq, S.; Rodriguez, J.; Beaumont, M.; Cani, P.D.; Bindels, L.B.; Lanthier, N.; et al. Hepatoprotective Effects of Indole, a Gut Microbial Metabolite, in Leptin-Deficient Obese Mice. J. Nutr. 2021, 151, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.D.; Palanivel, R.; Mottillo, E.P.; Bujak, A.L.; Wang, H.; Ford, R.J.; Collins, A.; Blumer, R.M.; Fullerton, M.D.; Yabut, J.M.; et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat. Med. 2015, 21, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Sumara, G.; Sumara, O.; Kim, J.K.; Karsenty, G. Gut-derived serotonin is a multifunctional determinant to fasting adaptation. Cell Metab. 2012, 16, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.H.; Park, B.L.; Kim, H.; German, M.S.; Go, M.J.; Jung, H.S.; Koo, B.K.; Cho, Y.M.; Choi, S.H.; Cho, Y.S.; et al. Association of variations in TPH1 and HTR2B with gestational weight gain and measures of obesity. Obesity 2012, 20, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Rahimi, A.; Ni, Z.; Said, H.; Subramanian, V.S. Disintegration of colonic epithelial tight junction in uremia: A likely cause of CKD-associated inflammation. Nephrol. Dial. Transplant. 2012, 27, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Bammens, B.; Evenepoel, P.; Verbeke, K.; Vanrenterghem, Y. Impairment of small intestinal protein assimilation in patients with end-stage renal disease: Extending the malnutrition-inflammation-atherosclerosis concept. Am. J. Clin. Nutr. 2004, 80, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Cupisti, A.; D’Alessandro, C.; Gesualdo, L.; Cosola, C.; Gallieni, M.; Egidi, M.F.; Fusaro, M. Non-Traditional Aspects of Renal Diets: Focus on Fiber, Alkali and Vitamin K1 Intake. Nutrients 2017, 9, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafra, D.; Barros, A.F.; Fouque, D. Dietary protein metabolism by gut microbiota and its consequences for chronic kidney disease patients. Future Microbiol. 2013, 8, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, G.; Shibata, K.; Takizawa, T.; Ikeda, Y.; Tokita, Y.; Umemura, S.; Tochikubo, O. Prevalence of constipation in continuous ambulatory peritoneal dialysis patients and comparison with hemodialysis patients. Am. J. Kidney Dis. 2002, 39, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Xu, K.Y.; Xia, G.H.; Lu, J.Q.; Chen, M.X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.W.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1445. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Xie, S.; Lv, D.; Wang, P.; He, H.; Zhang, T.; Zhou, Y.; Lin, Q.; Zhou, H.; Jiang, J.; et al. Alteration of the gut microbiota in Chinese population with chronic kidney disease. Sci. Rep. 2017, 7, 2870. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef] [PubMed]
- Banoglu, E.; Jha, G.G.; King, R.S. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Evenepoel, P. The gut-kidney axis: Indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol. Dial. Transplant. 2011, 26, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Meyer, T.W.; Hostetter, T.H.; Melamed, M.L.; Parekh, R.S.; Hwang, S.; Banerjee, T.; Coresh, J.; Powe, N.R. Free Levels of Selected Organic Solutes and Cardiovascular Morbidity and Mortality in Hemodialysis Patients: Results from the Retained Organic Solutes and Clinical Outcomes (ROSCO) Investigators. PLoS ONE 2015, 10, e0126048. [Google Scholar] [CrossRef]
- Melamed, M.L.; Plantinga, L.; Shafi, T.; Parekh, R.; Meyer, T.W.; Hostetter, T.H.; Coresh, J.; Powe, N.R. Retained organic solutes, patient characteristics and all-cause and cardiovascular mortality in hemodialysis: Results from the retained organic solutes and clinical outcomes (ROSCO) investigators. BMC Nephrol. 2013, 14, 134. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Yang, S.T.; Li, S.H.; Zhao, L.; Hao, Y.L.; Qin, J.J.; Zhang, L.; Zhang, C.Y.; Bian, W.J.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Hsu, H.H.; Wu, M.S. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol. Dial. Transplant. 2013, 28, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Muteliefu, G.; Enomoto, A.; Nishijima, F.; Dateki, M.; Niwa, T. NF-kappaB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2011, 301, C1201–C1212. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar]
- Lobel, L.; Cao, Y.G.; Fenn, K.; Glickman, J.N.; Garrett, W.S. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 2020, 369, 1518–1524. [Google Scholar] [CrossRef]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef]
- Lano, G.; Laforet, M.; Von Kotze, C.; Perrin, J.; Addi, T.; Brunet, P.; Poitevin, S.; Burtey, S.; Dou, L. Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Metabolic Disorders | |||
| Metabolite | Increased/Decreased/Association | Patient Group | Reference |
| Kynurenine/tryptophan ratio | Increased | Obese/metabolic syndrome | [76] |
| Kynurenine | + associated with BMI, HOMA-index | Diabetes | [147] |
| Kynurenine | Increased | Type 2 diabetes/obese | [148] |
| Indole | Decreased (feces) | Type 2 diabetes/obese | [149] |
| Indole-3 acetic acid | Decreased (feces) | Type 2 diabetes/obese | [149] |
| 3-methyl-indole | Decreased (feces) | Type 2 diabetes/obese | [149] |
| Tryptamine | Decreased (feces) | Type 2 diabetes/obese | [149] |
| Indole-3 propionic acid | + associated with insulin sensitivity | Type 2 diabetes | [150] |
| Indole-3 propionic acid | Decreased | Type 2 diabetes/obese | [94] |
| 5-hydroxyindole-3-acetic acid | Increased | Metabolic syndrome | [151] |
| Chronic Kidney and Cardiovascular Diseases | |||
| Metabolite | Increased/Decreased/Association | Patient Group | Reference |
| Indoxyl sulfate | Increased | CKD, ESRD | [32,152] |
| Indoxyl sulfate | + associated with mortality | CKD | [153] |
| Indoxyl sulfate | + associated with hearth failure | CKD hemodyalysis | [154] |
| Indoxyl sulfate | + associated with aortic calcification | CKD | [155] |
| Indoxyl sulfate | + associated with arterial stiffness | CKD | [155] |
| Kynurenine | + associated with myocardial infarction | Coronary artery disease | [71] |
| Kynurenine | Increased (in unstable atherosclerotic plaques) | Coronary artery disease | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Nieuwdorp, M.; de Vos, W.M.; Rampanelli, E. Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders. Metabolites 2022, 12, 834. https://doi.org/10.3390/metabo12090834
Liu M, Nieuwdorp M, de Vos WM, Rampanelli E. Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders. Metabolites. 2022; 12(9):834. https://doi.org/10.3390/metabo12090834
Chicago/Turabian StyleLiu, Moyan, Max Nieuwdorp, Willem M. de Vos, and Elena Rampanelli. 2022. "Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders" Metabolites 12, no. 9: 834. https://doi.org/10.3390/metabo12090834
APA StyleLiu, M., Nieuwdorp, M., de Vos, W. M., & Rampanelli, E. (2022). Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders. Metabolites, 12(9), 834. https://doi.org/10.3390/metabo12090834