
Prostanoid Metabolites as Biomarkers in Human Disease


Abstract
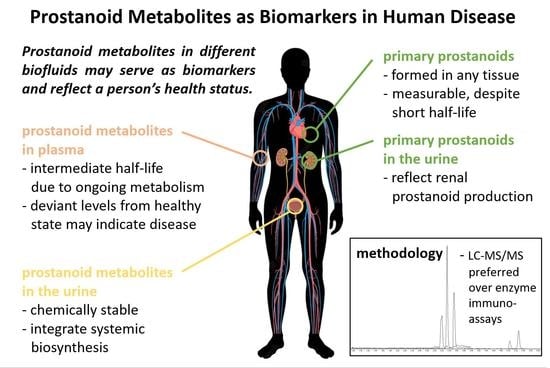
1. Introduction
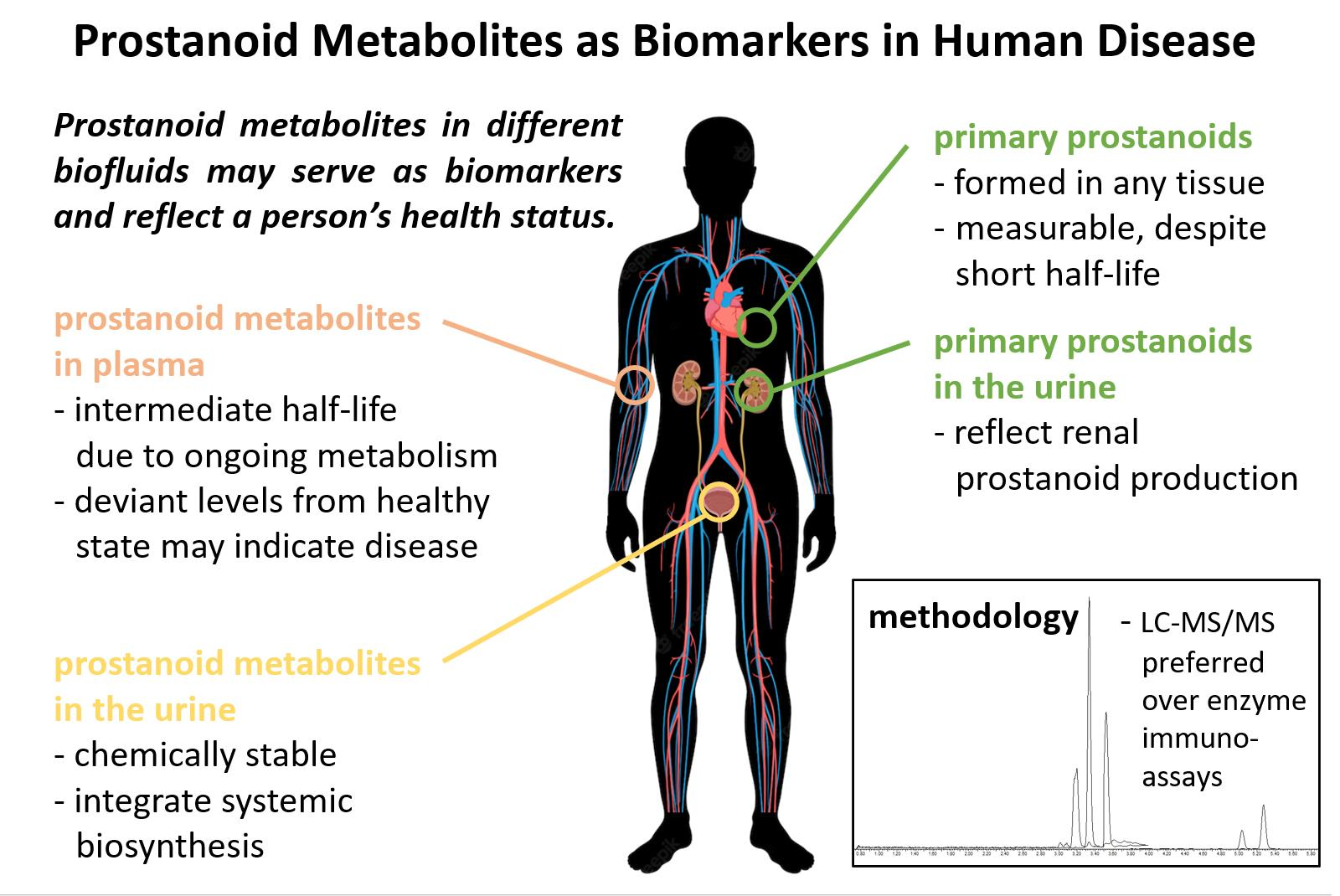
2. Metabolism of Prostanoids
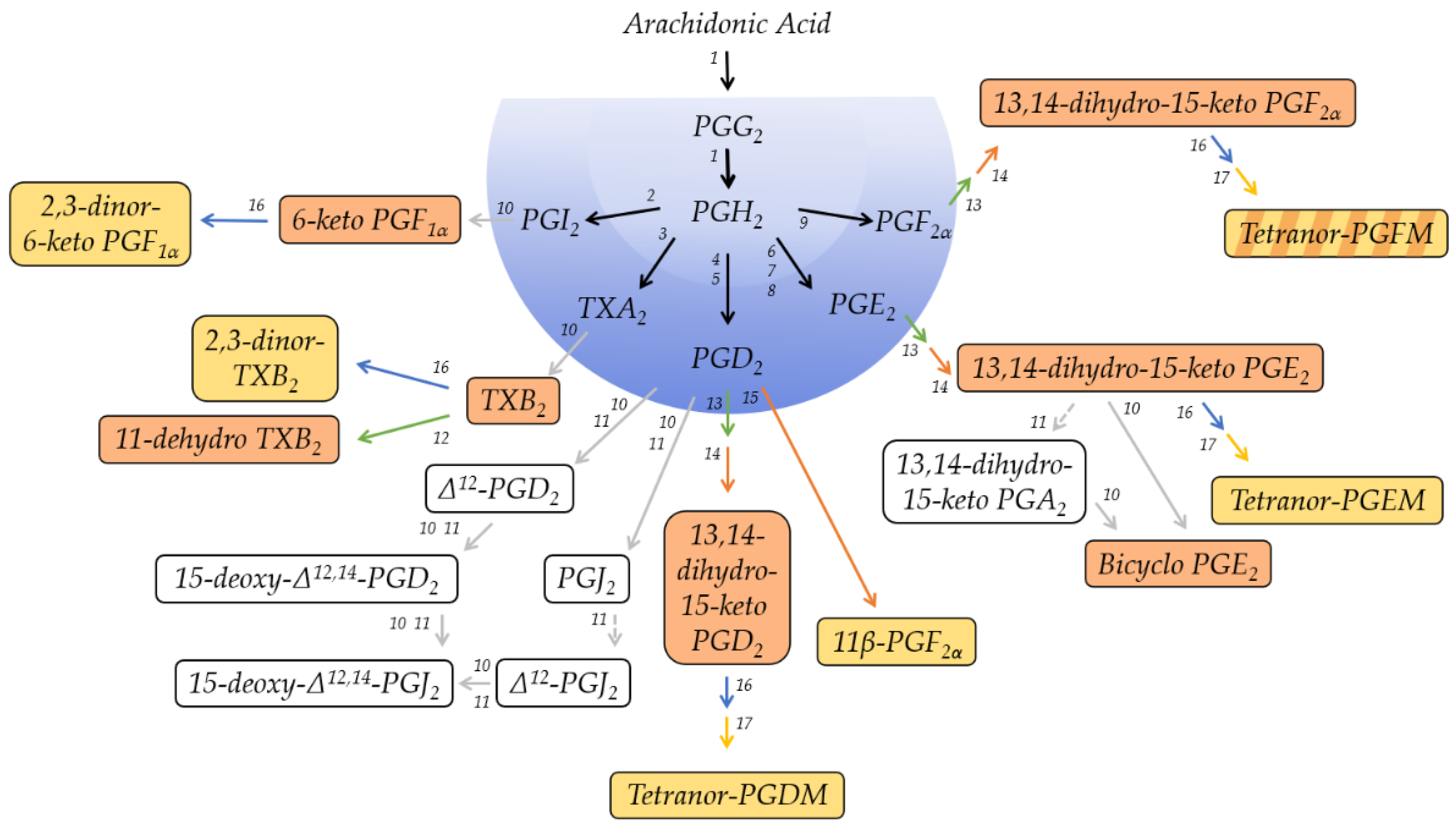
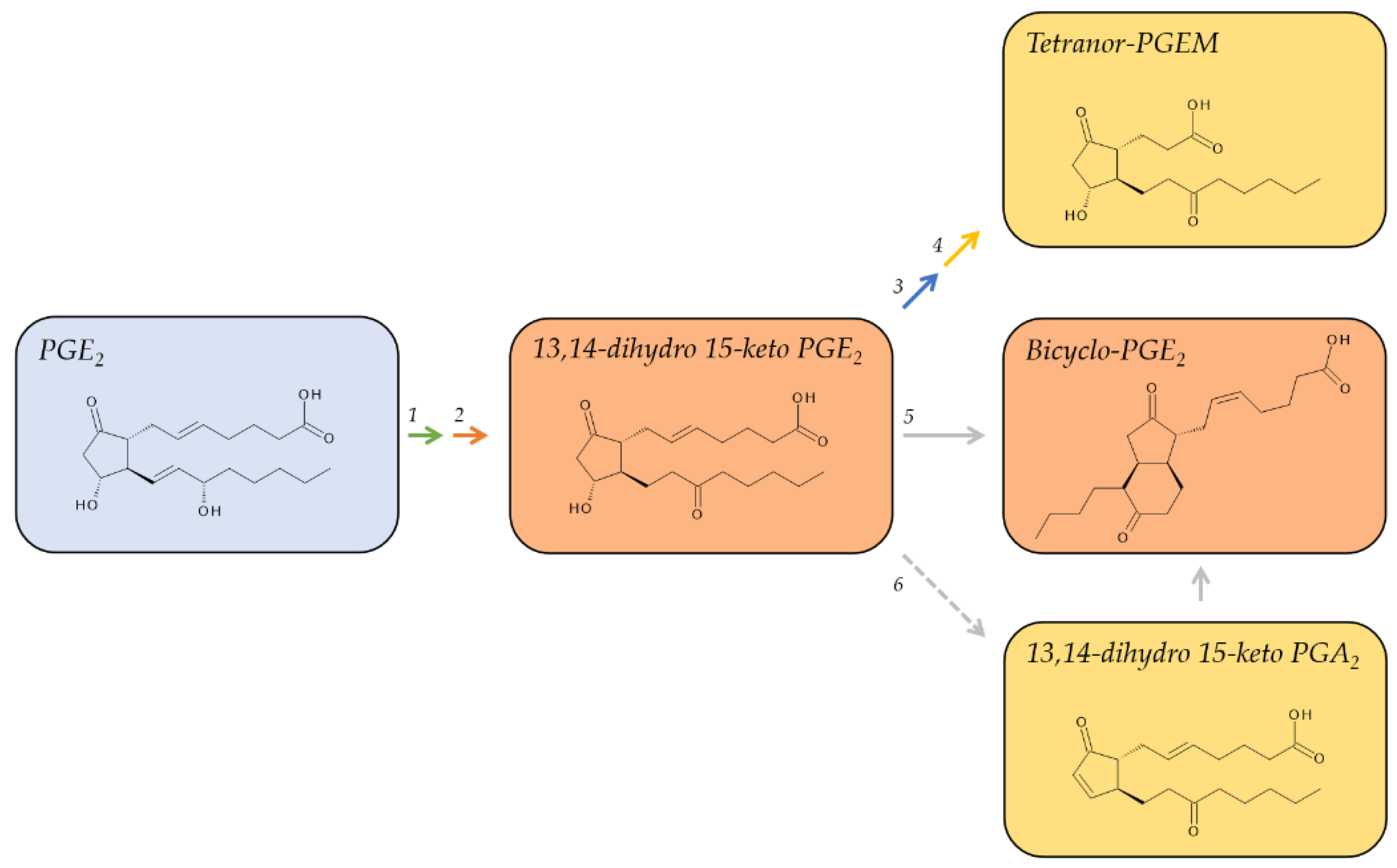
2.1. Metabolites of PGE2
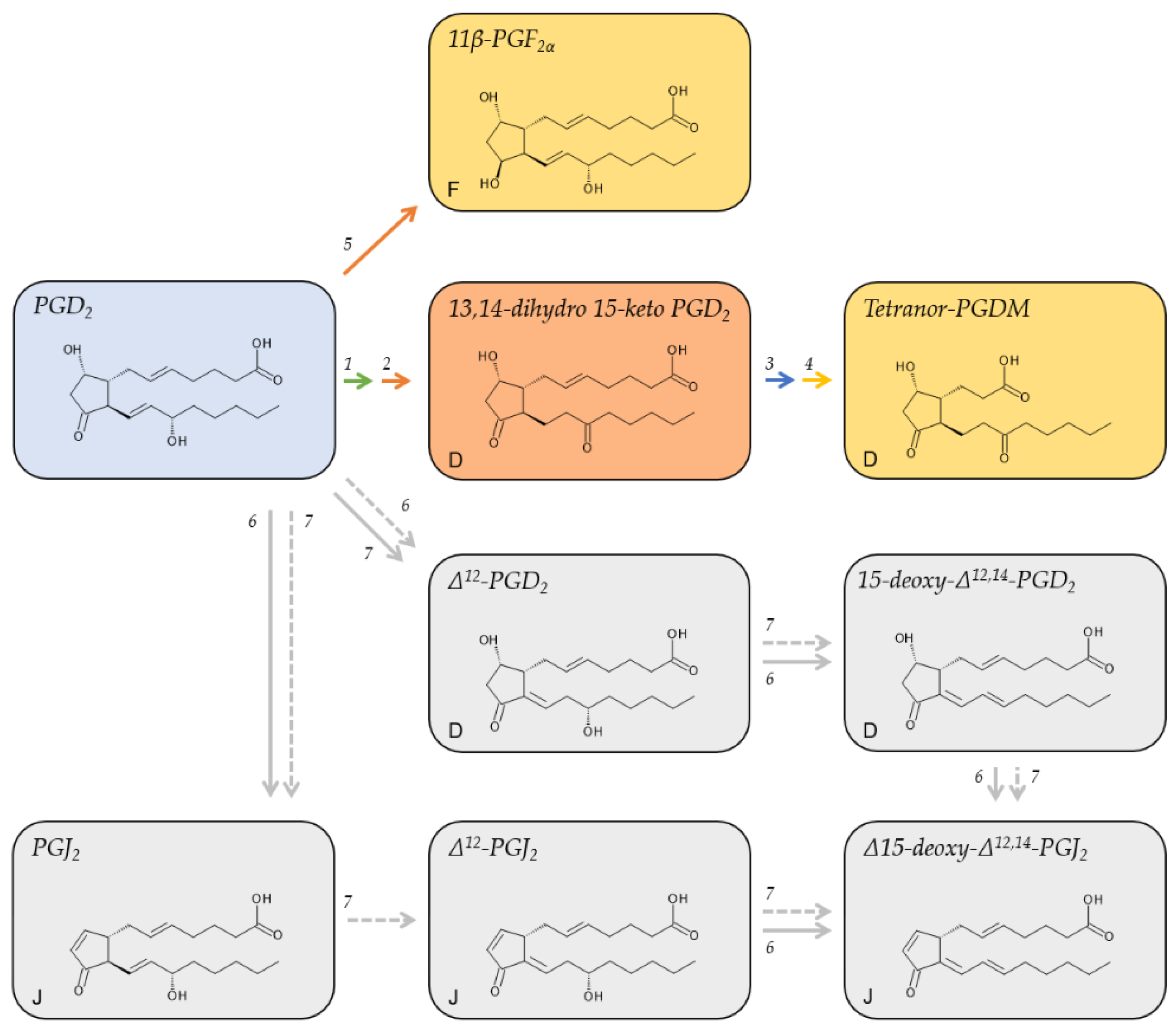
2.2. Metabolites of PGD2
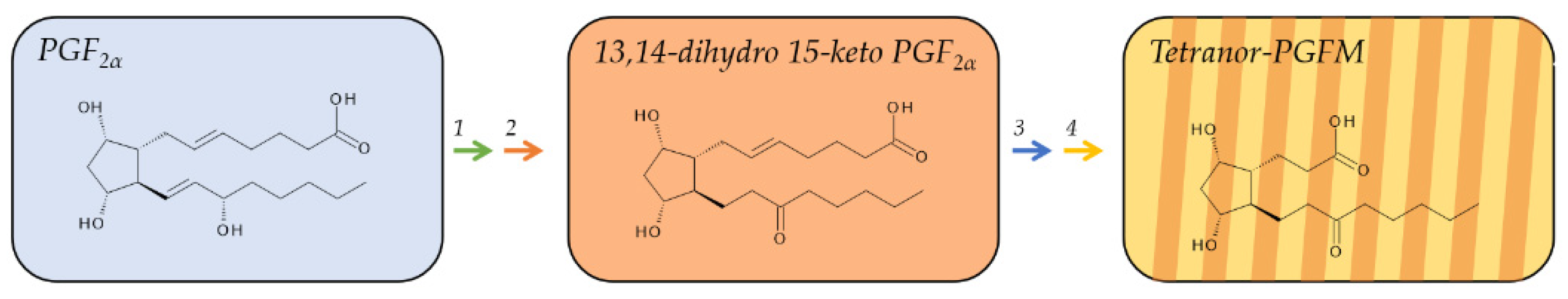
2.3. Metabolites of PGF2α
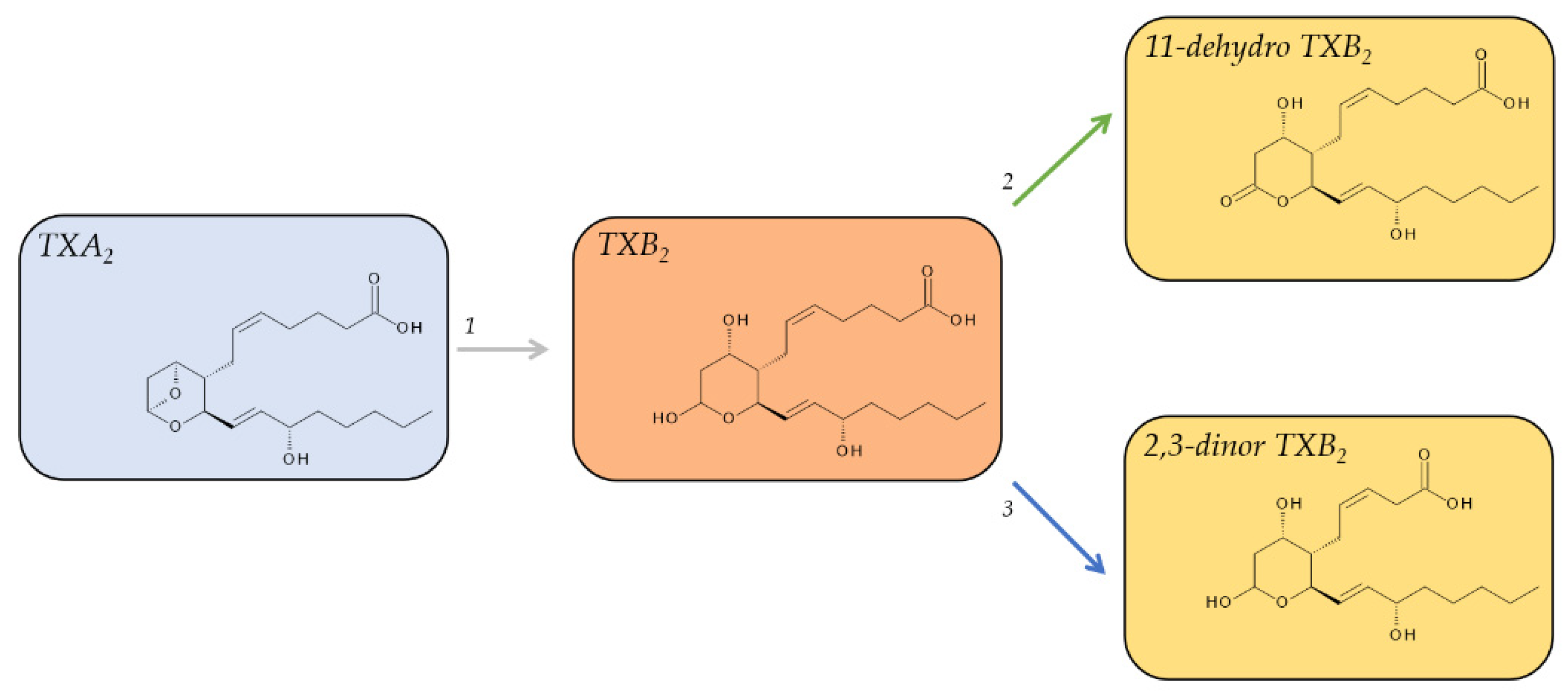
2.4. Metabolites of TXA2
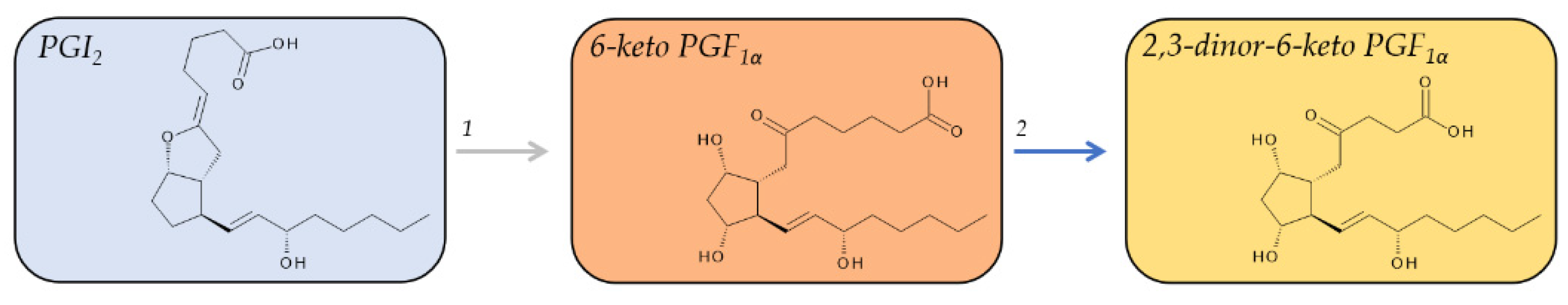
2.5. Metabolites of PGI2
3. Analysis of Prostanoid Metabolites
3.1. Sample Selection and Choice of Analyte(s)
3.2. Sample Collection, Handling, and Storage
3.3. Methodology for the Analysis of Prostanoid Lipid Mediators
4. Frequently Encountered Obstacles When Analyzing Prostanoid Metabolites
4.1. Accuracy of Analytical Results Obtained by Immunoassay versus LC–MS/MS
4.2. Undisclosed Use of NSAIDs May Compromise Study Results
4.3. Normalization of Urinary Metabolites
4.4. Collection of 24 h Urine versus Spot Urine Samples
4.5. Renal versus Extrarenal Origin of Urinary Prostanoid Metabolites and Data Interpretation
5. Prostanoid Metabolites as Relevant Biomarkers in Human Disease
5.1. PGE2 Metabolites as Biomarkers
5.2. PGD2 Metabolites as Biomarkers
5.3. PGF2α Metabolites as Biomarkers
5.4. TXA2 Metabolites as Biomarkers
5.5. PGI2 Metabolites as Biomarkers
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitchell, J.A.; Kirkby, N.S.; Ahmetaj-Shala, B.; Armstrong, P.C.; Crescente, M.; Ferreira, P.; Lopes Pires, M.E.; Vaja, R.; Warner, T.D. Cyclooxygenases and the cardiovascular system. Pharmacol. Ther. 2021, 217, 107624. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, B.; Gowda, S.; Welty, S.E.; Barrington, K.J.; Pammi, M. Prostanoids and their analogues for the treatment of pulmonary hypertension in neonates. Cochrane Database Syst. Rev. 2019, 10, CD012963. [Google Scholar] [CrossRef]
- Yang, T.; Liu, M. Regulation and function of renal medullary cyclooxygenase-2 during high salt loading. Front. Biosci. 2017, 22, 128–136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahmad, A.S.; Ottallah, H.; Maciel, C.B.; Strickland, M.; Doré, S. Role of the L-PGDS-PGD2-DP1 receptor axis in sleep regulation and neurologic outcomes. Sleep 2019, 42, zsz073. [Google Scholar] [CrossRef] [PubMed]
- Urade, Y.; Hayaishi, O. Prostaglandin D2 and sleep/wake regulation. Sleep Med. Rev. 2011, 15, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.M.; Hornaday, K.K.; Slater, D.M. Prostaglandins in biofluids in pregnancy and labour: A systematic review. PLoS ONE 2021, 16, e0260115. [Google Scholar] [CrossRef] [PubMed]
- Garami, A.; Steiner, A.A.; Romanovsky, A.A. Fever and hypothermia in systemic inflammation. Handb. Clin. Neurol. 2018, 157, 565–597. [Google Scholar] [CrossRef] [PubMed]
- Idborg, H.; Pawelzik, S.C.; Perez-Manso, M.; Bjork, L.; Hamrin, J.; Herlenius, E.; Jakobsson, P.J. Evaluation of urinary prostaglandin E2 metabolite as a biomarker in infants with fever due to viral infection. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Engström, L.; Mackerlova, L.; Jakobsson, P.J.; Blomqvist, A. Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1100–R1107. [Google Scholar] [CrossRef]
- Kobayashi, K.; Omori, K.; Murata, T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. 2018, 37, 347–354. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J. Neuroinflamm. 2020, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Grösch, S.; Niederberger, E.; Geisslinger, G. Investigational drugs targeting the prostaglandin E2 signaling pathway for the treatment of inflammatory pain. Expert Opin. Investig. Drugs 2017, 26, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Leclerc, P.; Pawelzik, S.C.; Idborg, H.; Spahiu, L.; Larsson, C.; Stenberg, P.; Korotkova, M.; Jakobsson, P.J. Characterization of a new mPGES-1 inhibitor in rat models of inflammation. Prostaglandins Other Lipid Mediat. 2013, 102–103, 1–12. [Google Scholar] [CrossRef]
- Wang, Q.; Morris, R.J.; Bode, A.M.; Zhang, T. Prostaglandin Pathways: Opportunities for Cancer Prevention and Therapy. Cancer Res. 2022, 82, 949–965. [Google Scholar] [CrossRef]
- Hanaka, H.; Pawelzik, S.C.; Johnsen, J.I.; Rakonjac, M.; Terawaki, K.; Rasmuson, A.; Sveinbjörnsson, B.; Schumacher, M.C.; Hamberg, M.; Samuelsson, B.; et al. Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18757–18762. [Google Scholar] [CrossRef]
- Biringer, R.G. The enzymology of the human prostanoid pathway. Mol. Biol. Rep. 2020, 47, 4569–4586. [Google Scholar] [CrossRef]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef]
- Biringer, R.G. A Review of Prostanoid Receptors: Expression, Characterization, Regulation, and Mechanism of Action. J. Cell Commun. Signal. 2020, 15, 155–184. [Google Scholar] [CrossRef]
- Smith, W.L.; Malkowski, M.G. Interactions of fatty acids, nonsteroidal anti-inflammatory drugs, and coxibs with the catalytic and allosteric subunits of cyclooxygenases-1 and -2. J. Biol. Chem. 2019, 294, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Van der Donk, W.A.; Tsai, A.L.; Kulmacz, R.J. The cyclooxygenase reaction mechanism. Biochemistry 2002, 41, 15451–15458. [Google Scholar] [CrossRef]
- Hamberg, M.; Samuelsson, B. On the mechanism of the biosynthesis of prostaglandins E-1 and F-1-alpha. J. Biol. Chem. 1967, 242, 5336–5343. [Google Scholar] [CrossRef]
- Hamberg, M.; Samuelsson, B. Oxygenation of unsaturated fatty acids by the vesicular gland of sheep. J. Biol. Chem. 1967, 242, 5344–5354. [Google Scholar] [CrossRef]
- Seo, M.J.; Oh, D.K. Prostaglandin synthases: Molecular characterization and involvement in prostaglandin biosynthesis. Prog. Lipid Res. 2017, 66, 50–68. [Google Scholar] [CrossRef]
- Sun, C.-C.; Zhou, Z.-Q.; Yang, D.; Chen, Z.-L.; Zhou, Y.-Y.; Wen, W.; Feng, C.; Zheng, L.; Peng, X.-Y.; Tang, C.-F. Recent advances in studies of 15-PGDH as a key enzyme for the degradation of prostaglandins. Int. Immunopharmacol. 2021, 101, 108176. [Google Scholar] [CrossRef] [PubMed]
- Hamberg, M.; Samuelsson, B. On the Metabolism of Prostaglandins E1 and E2 in Man. J. Biol. Chem. 1971, 246, 6713–6721. [Google Scholar] [CrossRef]
- Bothwell, W.; Verburg, M.; Wynalda, M.; Daniels, E.G.; Fitzpatrick, F.A. A radioimmunoassay for the unstable pulmonary metabolites of prostaglandin E1 and E2: An indirect index of their in vivo disposition and pharmacokinetics. J. Pharmacol. Exp. Ther. 1982, 220, 229–235. [Google Scholar]
- Fitzpatrick, F.A.; Aguirre, R.; Pike, J.E.; Lincoln, F.H. The stability of 13,14-dihydro-15 keto-PGE2. Prostaglandins 1980, 19, 917–931. [Google Scholar] [CrossRef]
- Pawelzik, S.C.; Avignon, A.; Idborg, H.; Boegner, C.; Stanke-Labesque, F.; Jakobsson, P.J.; Sultan, A.; Back, M. Urinary prostaglandin D2 and E2 metabolites associate with abdominal obesity, glucose metabolism, and triglycerides in obese subjects. Prostaglandins Other Lipid Mediat. 2019, 145, 106361. [Google Scholar] [CrossRef]
- Leonhardt, A.; Krauss, M.; Gieler, U.; Schweer, H.; Happle, R.; Seyberth, H.W. In vivo formation of prostaglandin E1 and prostaglandin E2 in atopic dermatitis. Br. J. Dermatol. 1997, 136, 337–340. [Google Scholar]
- Schuligoi, R.; Schmidt, R.; Geisslinger, G.; Kollroser, M.; Peskar, B.A.; Heinemann, A. PGD2 metabolism in plasma: Kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochem. Pharmacol. 2007, 74, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.; Fleming, T.; Kadiyska, I.; Brings, S.; Groener, J.B.; Nawroth, P.; Hecker, M.; Brune, M. Sensitive mass spectrometric assay for determination of 15-deoxy-Δ12,14-prostaglandin J2 and its application in human plasma samples of patients with diabetes. Anal. Bioanal. Chem. 2017, 410, 521–528. [Google Scholar] [CrossRef]
- Bell-Parikh, L.C.; Ide, T.; Lawson, J.A.; McNamara, P.; Reilly, M.; FitzGerald, G.A. Biosynthesis of 15-deoxy-Δ12,14-PGJ2 and the ligation of PPARγ. J. Clin. Investig. 2003, 112, 945–955. [Google Scholar] [CrossRef]
- Meyer, H.H.; Eisele, K.; Osaso, J. A biotin-streptavidin amplified enzymeimmunoassay for 13,14-dihydro-15-keto-PGF2 alpha. Prostaglandins 1989, 38, 375–383. [Google Scholar] [CrossRef]
- Granström, E.; Kindahl, H.; Swahn, M.L. Profiles of prostaglandin metabolites in the human circulation. Identification of late-appearing, long-lived products. Biochim. Biophys. Acta 1982, 713, 46–60. [Google Scholar] [CrossRef]
- Medina, S.; Domínguez-Perles, R.; Gil, J.I.; Ferreres, F.; García-Viguera, C.; Martínez-Sanz, J.M.; Gil-Izquierdo, A. A ultra-pressure liquid chromatography/triple quadrupole tandem mass spectrometry method for the analysis of 13 eicosanoids in human urine and quantitative 24 hour values in healthy volunteers in a controlled constant diet. Rapid Commun. Mass Spectrom. 2012, 26, 1249–1257. [Google Scholar] [CrossRef]
- Needleman, P.; Moncada, S.; Bunting, S.; Vane, J.R.; Hamberg, M.; Samuelsson, B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature 1976, 261, 558–560. [Google Scholar] [CrossRef]
- Patrono, C.; Rocca, B. Measurement of Thromboxane Biosynthesis in Health and Disease. Front. Pharmacol. 2019, 10, 1244. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Ciabattoni, G.; Pugliese, F.; Pierucci, A.; Blair, I.A.; FitzGerald, G.A. Estimated rate of thromboxane secretion into the circulation of normal humans. J. Clin. Investig. 1986, 77, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Catella, F.; Healy, D.; Lawson, J.A.; FitzGerald, G.A. 11-Dehydrothromboxane B2: A quantitative index of thromboxane A2 formation in the human circulation. Proc. Natl. Acad. Sci. USA 1986, 83, 5861–5865. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Davì, G.; Basili, S.; Lattanzio, S.; Cavoni, A.; Guizzardi, G.; De Feudis, L.; Traisci, G.; Pettinella, C.; Paloscia, L.; et al. Thromboxane and prostacyclin biosynthesis in heart failure of ischemic origin: Effects of disease severity and aspirin treatment. J. Thromb. Haemost. 2010, 8, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Machleidt, C.; Förstermann, U.; Anhut, H.; Hertting, G. Formation and elimination of prostacyclin metabolites in the cat in vivo as determined by radioimmunoassay of unextracted plasma. Eur. J. Pharmacol. 1981, 74, 19–26. [Google Scholar] [CrossRef]
- Orchard, M.A.; Robinson, C. Stability of prostacyclin in human plasma and whole blood: Studies on the protective effect of albumin. Thromb. Haemost. 1981, 46, 645–647. [Google Scholar] [CrossRef]
- Polterauer, P.; Sinzinger, H.; Peskar, B.A. Biological half-life of prostacyclin and 6-oxo-PGF1 alpha levels in plasma of patients with colonic cancer. Prostaglandins Leukot. Med. 1986, 22, 249–258. [Google Scholar] [CrossRef]
- Lucas, F.V.; Skrinska, V.A.; Chisolm, G.M.; Hesse, B.L. Stability of prostacyclin in human and rabbit whole blood and plasma. Thromb. Res. 1986, 43, 379–387. [Google Scholar] [CrossRef]
- Chappell, D.L.; Xiao, X.; Radziszewski, W.; Laterza, O.F. Development and validation of a LC/MS/MS method for 6-keto PGF1α, a metabolite of prostacyclin (PGI2). J. Pharm. Biomed. Anal. 2011, 56, 600–603. [Google Scholar] [CrossRef]
- Wennmalm, A.; Benthin, G.; Granström, E.F.; Persson, L.; Winell, S. 2,3-Dinor metabolites of thromboxane A2 and prostacyclin in urine from healthy human subjects: Diurnal variation and relation to 24h excretion. Clin. Sci. 1992, 83, 461–465. [Google Scholar] [CrossRef]
- Liston, T.E.; Roberts, L.J., 2nd. Metabolic fate of radiolabeled prostaglandin D2 in a normal human male volunteer. J. Biol. Chem. 1985, 260, 13172–13180. [Google Scholar] [CrossRef]
- Roberts, L.J., 2nd; Sweetman, B.J.; Oates, J.A. Metabolism of thromboxane B2 in man. Identification of twenty urinary metabolites. J. Biol. Chem. 1981, 256, 8384–8393. [Google Scholar] [CrossRef]
- Bygdeman, M. Pharmacokinetics of prostaglandins. Best Pract. Res. Clin. Obstet. Gynaecol. 2003, 17, 707–716. [Google Scholar] [CrossRef]
- Chou, W.L.; Chuang, L.M.; Chou, C.C.; Wang, A.H.; Lawson, J.A.; FitzGerald, G.A.; Chang, Z.F. Identification of a novel prostaglandin reductase reveals the involvement of prostaglandin E2 catabolism in regulation of peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2007, 282, 18162–18172. [Google Scholar] [CrossRef] [PubMed]
- Granström, E.; Hamberg, M.; Hansson, G.; Kindahl, H. Chemical instability of 15-keto-13,14-dihydro-PGE 2: The reason for low assay reliability. Prostaglandins 1980, 19, 933–957. [Google Scholar] [CrossRef]
- Murphy, R.C.; FitzGerald, G.A. Current approaches to estimation of eicosanoid formation in vivo. Adv. Prostaglandin Thromboxane Leukot. Res. 1994, 22, 341–348. [Google Scholar] [PubMed]
- Hamberg, M. Inhibition of prostaglandin synthesis in man. Biochem. Biophys. Res. Commun. 1972, 49, 720–726. [Google Scholar] [CrossRef]
- Honda, H.; Fukawa, K.; Sawabe, T. Influence of adjuvant arthritis on main urinary metabolites of prostaglandin F and E in rats. Prostaglandins 1980, 19, 259–269. [Google Scholar] [CrossRef]
- Song, W.-L.; Wang, M.; Ricciotti, E.; Fries, S.; Yu, Y.; Grosser, T.; Reilly, M.; Lawson, J.A.; FitzGerald, G.A. Tetranor PGDM, an Abundant Urinary Metabolite Reflects Biosynthesis of Prostaglandin D2 in Mice and Humans. J. Biol. Chem. 2008, 283, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, P.K.; Betti, P.A. Biological activity of metabolites of PGD2 on canine proximal colon. Am. J. Physiol. 1993, 264, G886–G894. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Spokas, E.G.; Marcinkiewicz, E.; Wong, P.Y. Hepatic transformation of prostaglandin D2 to a new prostanoid, 9 alpha, 11 beta-prostaglandin F2, that inhibits platelet aggregation and constricts blood vessels. J. Biol. Chem. 1985, 260, 14621–14625. [Google Scholar] [CrossRef]
- Seibert, K.; Sheller, J.R.; Roberts, L.J., 2nd. (5Z,13E)-(15S)-9 alpha, 11 beta,15-trihydroxyprosta-5,13-dien-1-oic acid (9 alpha, 11 beta-prostaglandin F2): Formation and metabolism by human lung and contractile effects on human bronchial smooth muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Liston, T.E.; Roberts, L.J., 2nd. Transformation of prostaglandin D2 to 9 alpha, 11 beta-(15S)-trihydroxyprosta-(5Z,13E)-dien-1-oic acid (9 alpha, 11 beta-prostaglandin F2): A unique biologically active prostaglandin produced enzymatically in vivo in humans. Proc. Natl. Acad. Sci. USA 1985, 82, 6030–6034. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Sheldrick, R.L.G. Prostanoid-induced contraction of human bronchial smooth muscle is mediated by TP-receptors. Br. J. Pharmacol. 1989, 96, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.J., 2nd; Seibert, K.; Liston, T.E.; Tantengco, M.V.; Robertson, R.M. PGD2 is transformed by human coronary arteries to 9 alpha, 11 beta-PGF2, which contracts human coronary artery rings. Adv. Prostaglandin Thromboxane Leukot. Res. 1987, 17A, 427–429. [Google Scholar] [PubMed]
- Yoda, T.; Kikuchi, K.; Miki, Y.; Onodera, Y.; Hata, S.; Takagi, K.; Nakamura, Y.; Hirakawa, H.; Ishida, T.; Suzuki, T.; et al. 11β-Prostaglandin F2α, a bioactive metabolite catalyzed by AKR1C3, stimulates prostaglandin F receptor and induces slug expression in breast cancer. Mol. Cell. Endocrinol. 2015, 413, 236–247. [Google Scholar] [CrossRef]
- Söderström, M.; Wigren, J.; Surapureddi, S.; Glass, C.K.; Hammarström, S. Novel prostaglandin D 2-derived activators of peroxisome proliferator-activated receptor-γ are formed in macrophage cell cultures. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2003, 1631, 35–41. [Google Scholar] [CrossRef]
- Fukushima, M.; Sasaki, H.; Fukushima, S. Prostaglandin J2 and Related Compounds. Ann. N. Y. Acad. Sci. 1994, 744, 161–165. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef]
- Straus, D.S.; Glass, C.K. Cyclopentenone prostaglandins: New insights on biological activities and cellular targets. Med. Res. Rev. 2001, 21, 185–210. [Google Scholar] [CrossRef]
- Levine, L.; Gutierrez-Cernosek, R.M. Levels of 13,14-dihydro-15-keto-PGF2α in biological fluids as measured by radioimmunoassay. Prostaglandins 1973, 3, 785–804. [Google Scholar] [CrossRef]
- Samuelsson, B.; Goldyne, M.; Granström, E.; Hamberg, M.; Hammarström, S.; Malmsten, C. Prostaglandins and thromboxanes. Annu. Rev. Biochem. 1978, 47, 997–1029. [Google Scholar] [CrossRef]
- Granstrom, E.; Samuelsson, B. Structure of a urinary metabolite of prostaglandin F2.alpha. in man. J. Am. Chem. Soc. 1969, 91, 3398–3400. [Google Scholar] [CrossRef]
- Neves, A.C.O.; Morais, C.L.M.; Mendes, T.P.P.; Vaz, B.G.; Lima, K.M.G. Mass spectrometry and multivariate analysis to classify cervical intraepithelial neoplasia from blood plasma: An untargeted lipidomic study. Sci. Rep. 2018, 8, 3910–3954. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B. Role of basic science in the development of new medicines: Examples from the eicosanoid field. J. Biol. Chem. 2012, 287, 10070–10080. [Google Scholar] [CrossRef] [PubMed]
- Westlund, P.; Fylling, A.C.; Cederlund, E.; Jörnvall, H. 11-Hydroxythromboxane B2 dehydrogenase is identical to cytosolic aldehyde dehydrogenase. FEBS Lett. 1994, 345, 99–103. [Google Scholar] [CrossRef]
- Westlund, P.; Granström, E.; Kumlin, M.; Nordenström, A. Identification of 11-dehydro-TXB2 as a suitable parameter for monitoring thromboxane production in the human. Prostaglandins 1986, 31, 929–960. [Google Scholar] [CrossRef]
- Lawson, J.A.; Brash, A.R.; Doran, J.; FitzGerald, G.A. Measurement of urinary 2,3-dinor-thromboxane B2 and thromboxane B2 using bonded-phase phenylboronic acid columns and capillary gas chromatography--negative-ion chemical ionization mass spectrometry. Anal. Biochem. 1985, 150, 463–470. [Google Scholar] [CrossRef]
- Lellouche, F.; Fradin, A.; Fitzgerald, G.; Maclouf, J. Enzyme immunoassay measurement of the urinary metabolites of thromboxane A2 and prostacyclin. Prostaglandins 1990, 40, 297–310. [Google Scholar] [CrossRef]
- Perneby, C.; Granström, E.; Beck, O.; Fitzgerald, D.; Harhen, B.; Hjemdahl, P. Optimization of an enzyme immunoassay for 11-dehydro-thromboxane B(2) in urine: Comparison with GC-MS. Thromb. Res. 1999, 96, 427–436. [Google Scholar] [CrossRef]
- Lewis, P.J.; Dollery, C.T. Clinical pharmacology and potential of prostacyclin. Br. Med. Bull. 1983, 39, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R.; Jackson, E.K.; Saggese, C.A.; Lawson, J.A.; Oates, J.A.; FitzGerald, G.A. Metabolic disposition of prostacyclin in humans. J. Pharmacol. Exp. Ther. 1983, 226, 78–87. [Google Scholar] [PubMed]
- Flavahan, N.A. Balancing prostanoid activity in the human vascular system. Trends Pharmacol. Sci. 2006, 28, 106–110. [Google Scholar] [CrossRef]
- Rosenkranz, B.; Fischer, C.; Reimann, I.; Weimer, K.E.; Beck, G.; Frölich, J.C. Identification of the major metabolite of prostacyclin and 6-ketoprostaglandin F1α in man. Biochim. Biophys. Acta Lipids Lipid Metab. 1980, 619, 207–213. [Google Scholar] [CrossRef]
- Patrono, C.; Dunn, M.J. The clinical significance of inhibition of renal prostaglandin synthesis. Kidney Int. 1987, 32, 1–12. [Google Scholar] [CrossRef]
- FitzGerald, G.A.; Brash, A.R.; Falardeau, P.; Oates, J.A. Estimated rate of prostacyclin secretion into the circulation of normal man. J. Clin. Investig. 1981, 68, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Dahl, S.R.; Kleiveland, C.R.; Kassem, M.; Lea, T.; Lundanes, E.; Greibrokk, T. Detecting pM concentrations of prostaglandins in cell culture supernatants by capillary SCX-LC-MS/MS. J. Sep. Sci. 2008, 31, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Matsunuma, S.; Handa, S.; Kamei, D.; Yamamoto, H.; Okuyama, K.; Kato, Y. Oxaliplatin induces prostaglandin E2 release in vascular endothelial cells. Cancer Chemother. Pharmacol. 2019, 84, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Popov, T.A.M.D.P. Human exhaled breath analysis. Ann. Allergy Asthma Immunol. 2011, 106, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Degousee, N.; Fazel, S.; Angoulvant, D.; Stefanski, E.; Pawelzik, S.C.; Korotkova, M.; Arab, S.; Liu, P.; Lindsay, T.F.; Zhuo, S.; et al. Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation 2008, 117, 1701–1710. [Google Scholar] [CrossRef]
- Catella, F.; Nowak, J.; Fitzgerald, G.A. Measurement of renal and non-renal eicosanoid synthesis. Am. J. Med. 1986, 81, 23–29. [Google Scholar] [CrossRef]
- Granström, E.; Kumlin, M. Assay of thromboxane production in biological systems: Reliability of TXB2 versus 11-dehydro-TXB2 as targets for measurement. Adv. Prostaglandin Thromboxane Leukot. Res. 1987, 17, 587–594. [Google Scholar]
- Patrignani, P.; Tacconelli, S.; Piazuelo, E.; Di Francesco, L.; Dovizio, M.; Sostres, C.; Marcantoni, E.; Guillem-Llobat, P.; Del Boccio, P.; Zucchelli, M.; et al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J. Thromb. Haemost. 2014, 12, 1320–1330. [Google Scholar] [CrossRef]
- Bergstroem, S.; Ryhage, R.; Samuelsson, B.; Sjoevall, J. Prostaglandins and related factors. 15. the structures of prostaglandin e1, f1-alpha, and f1-beta. J. Biol. Chem. 1963, 238, 3555–3564. [Google Scholar] [PubMed]
- Blair, I.A. Measurement of eicosanoids by gas chromatography and mass spectrometry. Br. Med. Bull. 1983, 39, 223–226. [Google Scholar] [CrossRef]
- Blair, I.A. Electron-capture negative-ion chemical ionization mass spectrometry of lipid mediators. Methods Enzymol. 1990, 187, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.; Gonzalez-Riano, C.; Barbas, C.; Kolmert, J.; Hyung Ryu, M.; Carlsten, C.; Dahlén, S.-E.; Wheelock, C.E. Quantitative metabolic profiling of urinary eicosanoids for clinical phenotyping. J. Lipid Res. 2019, 60, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, Y.S.; Kanvinde, S.; Ahmad, R.; Singh, A.B.; Oupický, D.; Murry, D.J. Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS. Metabolites 2021, 11, 106. [Google Scholar] [CrossRef]
- Zhang, T.-Q.; Kuroda, H.; Nagano, K.; Terada, S.; Gao, J.-Q.; Harada, K.; Hirata, K.; Tsujino, H.; Higashisaka, K.; Matsumoto, H.; et al. Development and evaluation of a simultaneous and efficient quantification strategy for final prostanoid metabolites in urine. Prostaglandins Leukot. Essent. Fat. Acids 2020, 157, 102032. [Google Scholar] [CrossRef] [PubMed]
- Brose, S.A.; Thuen, B.T.; Golovko, M.Y. LC/MS/MS method for analysis of E2 series prostaglandins and isoprostanes. J. Lipid Res. 2011, 52, 850–859. [Google Scholar] [CrossRef]
- Balgoma, D.; Larsson, J.; Rokach, J.; Lawson, J.A.; Daham, K.; Dahlén, B.; Dahlén, S.-E.; Wheelock, C.E. Quantification of Lipid Mediator Metabolites in Human Urine from Asthma Patients by Electrospray Ionization Mass Spectrometry: Controlling Matrix Effects. Anal. Chem. 2013, 85, 7866–7874. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Peralbo, M.A.; de Castro, M.D.L. Preparation of urine samples prior to targeted or untargeted metabolomics mass-spectrometry analysis. Trac-Trends Anal. Chem. 2012, 41, 75–85. [Google Scholar] [CrossRef]
- Il’yasova, D.; Morrow, J.D.; Ivanova, A.; Wagenknecht, L.E. Epidemiological marker for oxidant status: Comparison of the ELISA and the gas chromatography/mass spectrometry assay for urine 2,3-dinor-5,6-dihydro-15-F2t-isoprostane. Ann. Epidemiol. 2004, 14, 793–797. [Google Scholar] [CrossRef]
- Mazaleuskaya, L.L.; Ricciotti, E. Druggable Prostanoid Pathway. Adv. Exp. Med. Biol. 2020, 1274, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fries, S.; Li, R.; Lawson, J.A.; Propert, K.J.; Diamond, S.L.; Blair, I.A.; FitzGerald, G.A.; Grosser, T. Differential impairment of aspirin-dependent platelet cyclooxygenase acetylation by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 16830–16835. [Google Scholar] [CrossRef] [PubMed]
- Khamis, M.M.; Holt, T.; Awad, H.; El-Aneed, A.; Adamko, D.J. Comparative analysis of creatinine and osmolality as urine normalization strategies in targeted metabolomics for the differential diagnosis of asthma and COPD. Metabolomics 2018, 14, 115. [Google Scholar] [CrossRef]
- Warrack, B.M.; Hnatyshyn, S.; Ott, K.H.; Reily, M.D.; Sanders, M.; Zhang, H.; Drexler, D.M. Normalization strategies for metabonomic analysis of urine samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 547–552. [Google Scholar] [CrossRef]
- Schurch, B.; Capaul, M.; Vallotton, M.B.; Rossier, A.B. Prostaglandin E2 measurements: Their value in the early diagnosis of heterotopic ossification in spinal cord injury patients. Arch. Phys. Med. Rehabil. 1997, 78, 687–691. [Google Scholar] [CrossRef]
- Remuzzi, G.; FitzGerald, G.A.; Patrono, C. Thromboxane synthesis and action within the kidney. Kidney Int. 1992, 41, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Bonvalet, J.P.; Pradelles, P.; Farman, N. Segmental synthesis and actions of prostaglandins along the nephron. Am. J. Physiol. 1987, 253, F377–F387. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Knowles, R.B.; Kirkby, N.S.; Reed, D.M.; Edin, M.L.; White, W.E.; Chan, M.V. Kidney Transplantation in a Patient Lacking Cytosolic Phospholipase A2 Proves Renal Origins of Urinary PGI-M and TX-M. Circ. Res. 2018, 122, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Knowles, R.B.; Kirkby, N.S.; Reed, D.M.; Edin, M.L.; White, W.E.; Chan, M.V.; Longhurst, H.; Yaqoob, M.M.; Milne, G.L.; et al. Letter by Mitchell et al. Regarding Article, “Urinary Prostaglandin Metabolites: An Incomplete Reckoning and a Flush to Judgment”. Circ. Res. 2018, 122, e84–e85. [Google Scholar] [CrossRef] [PubMed]
- Grosser, T.; Naji, A.; FitzGerald, G.A. Urinary Prostaglandin Metabolites: An Incomplete Reckoning and a Flush to Judgment. Circ. Res. 2018, 122, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, G.; Grosser, T. Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat. 2013, 104–105, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.W.; Geiman, D.E.; Hubbard, W.C.; Spannhake, E.W.; Hylind, L.M.; Hamilton, S.R.; Giardiello, F.M. Tissue prostanoids as biomarkers for chemoprevention of colorectal neoplasia: Correlation between prostanoid synthesis and clinical response in familial adenomatous polyposis. Prostaglandins Other Lipid Mediat. 2000, 60, 83–96. [Google Scholar] [CrossRef]
- Almer, G.; Teismann, P.; Stevic, Z.; Halaschek-Wiener, J.; Deecke, L.; Kostic, V.; Przedborski, S. Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology 2002, 58, 1277–1279. [Google Scholar] [CrossRef]
- Björk, L.; Leifsdottir, K.; Saha, S.; Herlenius, E. PGE2—Metabolite levels in CSF correlate to HIE score and outcome after perinatal asphyxia. Acta Paediatr. 2013, 102, 1041–1047. [Google Scholar] [CrossRef]
- Mattsson, N.; Yaong, M.; Rosengren, L.; Blennow, K.; Månsson, J.E.; Andersen, O.; Zetterberg, H.; Haghighi, S.; Zho, I.; Pratico, D. Elevated cerebrospinal fluid levels of prostaglandin E2 and 15-(S)-hydroxyeicosatetraenoic acid in multiple sclerosis. J. Intern. Med. 2009, 265, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Matsuura, T.; Matsuura, M.; Fujiwara, M.; Okayasu, I.; Ito, S.; Arihiro, S. Prostaglandin E-Major Urinary Metabolite as a Biomarker for Inflammation in Ulcerative Colitis: Prostaglandins Revisited. Digestion 2016, 93, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Campbell, J.; Yoo, W.; Taylor, J.A.; Sandler, D.P. Systemic levels of estrogens and PGE2 synthesis in relation to postmenopausal breast cancer risk. Cancer Epidemiol. Biomark. Prev. 2017, 26, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.C.; Schmidt, C.R.; Shrubsole, M.J.; Billheimer, D.D.; Joshi, P.R.; Morrow, J.D.; Heslin, M.J.; Washington, M.K.; Ness, R.M.; Zheng, W.; et al. Urine PGE-M: A metabolite of prostaglandin E2 as a potential biomarker of advanced colorectal neoplasia. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2006, 4, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Jabr, S.; Gartner, S.; Milne, G.L.; Roca-Ferrer, J.; Casas, J.; Moreno, A.; Gelpí, E.; Picado, C. Quantification of major urinary metabolites of PGE2 and PGD2 in cystic fibrosis: Correlation with disease severity. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 121–126. [Google Scholar] [CrossRef]
- Labat, C.; Temmar, M.; Nagy, E.; Bean, K.; Brink, C.; Benetos, A.; Bäck, M. Inflammatory mediators in saliva associated with arterial stiffness and subclinical atherosclerosis. J. Hypertens. 2013, 31, 2251–2258. [Google Scholar] [CrossRef] [PubMed]
- Anyona, S.B.; Kempaiah, P.; Davenport, G.C.; Vulule, J.M.; Hittner, J.B.; Ong’echa, J.M.; Perkins, D.J. Suppressed circulating bicyclo-PGE2 levels and leukocyte COX-2 transcripts in children co-infected with P. falciparum malaria and HIV-1 or bacteremia. Biochem. Biophys. Res. Commun. 2013, 436, 585–590. [Google Scholar] [CrossRef]
- Maeda, S.; Nakamura, T.; Harada, H.; Tachibana, Y.; Aritake, K.; Shimosawa, T.; Yatomi, Y.; Murata, T. Prostaglandin D2 metabolite in urine is an index of food allergy. Sci. Rep. 2017, 7, 17687. [Google Scholar] [CrossRef]
- Higashi, N.; Taniguchi, M.; Mita, H.; Yamaguchi, H.; Ono, E.; Akiyama, K. Aspirin-Intolerant Asthma (AIA) Assessment Using the Urinary Biomarkers, Leukotriene E4 (LTE4) and Prostaglandin D2 (PGD2) Metabolites. Allergol. Int. 2012, 61, 393–403. [Google Scholar] [CrossRef]
- Kolmert, J.; Gómez, C.; Balgoma, D.; Sjödin, M.; Bood, J.; Konradsen, J.R.; Ericsson, M.; Thörngren, J.-O.; James, A.; Mikus, M.; et al. Urinary Leukotriene E4 and Prostaglandin D2 Metabolites Increase in Adult and Childhood Severe Asthma Characterized by Type-2 Inflammation. Am. J. Respir. Crit. Care Med. 2020, 203, 37–53. [Google Scholar] [CrossRef]
- Takeshita, E.; Komaki, H.; Tachimori, H.; Miyoshi, K.; Yamamiya, I.; Shimizu-Motohashi, Y.; Ishiyama, A.; Saito, T.; Nakagawa, E.; Sugai, K.; et al. Urinary prostaglandin metabolites as Duchenne muscular dystrophy progression markers. Brain Dev. 2018, 40, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Buchheit, K.M.M.D.; Cahill, K.N.M.D.; Katz, H.P.; Murphy, K.; Feng, C.M.D.; Lee-Sarwar, K.; Lai, J.; Bhattacharyya, N.M.D.; Israel, E.M.D.F.; Boyce, J.A.M.D.F.; et al. Thymic Stromal Lymphopoietin Controls Prostaglandin D2 Generation in Aspirin-Exacerbated Respiratory Disease. J. Allergy Clin. Immunol. 2016, 137, AB198. [Google Scholar] [CrossRef][Green Version]
- Loomba, R.; Quehenberger, O.; Armando, A.; Dennis, E.A. Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis1. J. Lipid Res. 2015, 56, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, M.; Eckermann, O.; Babina, M.; Edenharter, G.; Worm, M. Serum levels of 9α,11β-PGF 2 and cysteinyl leukotrienes are useful biomarkers of anaphylaxis. J. Allergy Clin. Immunol. 2016, 137, 312–314.e7. [Google Scholar] [CrossRef]
- Coras, R.; Kavanaugh, A.; Kluzniak, A.; Holt, D.; Weilgosz, A.; Aaron, A.; Quehenberger, O.; Ritchlin, C.; Guma, M. Differences in oxylipin profile in psoriasis versus psoriatic arthritis. Arthritis Res. Ther. 2021, 23, 200. [Google Scholar] [CrossRef]
- Vazzana, N.; Santilli, F.; Lattanzio, S.; Liani, M.; Giacci, L.; Del Rosso, G.; Salvati, F.; Boccatonda, A.; Ferroni, P.; Davì, G. Determinants of thromboxane biosynthesis in patients with moderate to severe chronic kidney disease. Eur. J. Intern. Med. 2016, 33, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Szczuko, M.; Koziol, I.; Kotlega, D.; Brodowski, J.; Drozd, A. The Role of Thromboxane in the Course and Treatment of Ischemic Stroke: Review. Int. J. Mol. Sci. 2021, 22, 11644. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Vendrov, K.C.; Simmons, B.P.; Schuck, R.N.; Stouffer, G.A.; Lee, C.R. Urinary 11-dehydro-thromboxane B2 levels are associated with vascular inflammation and prognosis in atherosclerotic cardiovascular disease. Prostaglandins Other Lipid Mediat. 2018, 134, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Hishinuma, T.; Koseki, Y.; Murai, Y.; Yamazaki, T.; Suzuki, K.; Mizugaki, M. Urinary thromboxane A2/prostacyclin balance reflects the pathological state of a diabetic. Prostaglandins Other Lipid Mediat. 1999, 58, 263–271. [Google Scholar] [CrossRef]
- Ujike-Omori, H.; Maeshima, Y.; Kinomura, M.; Tanabe, K.; Mori, K.; Watatani, H.; Hinamoto, N.; Sugiyama, H.; Sakai, Y.; Morimatsu, H.; et al. The urinary levels of prostanoid metabolites predict acute kidney injury in heterogeneous adult Japanese ICU patients: A prospective observational study. Clin. Exp. Nephrol. 2015, 19, 1024–1036. [Google Scholar] [CrossRef]
- Thorén, S.; Jakobsson, P.J. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur. J. Biochem. 2000, 267, 6428–6434. [Google Scholar] [CrossRef]
- Stichtenoth, D.O.; Thorén, S.; Bian, H.; Peters-Golden, M.; Jakobsson, P.J.; Crofford, L.J. Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J. Immunol. 2001, 167, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Qasem, H.; Al-Ayadhi, L.; Bjørklund, G.; Chirumbolo, S.; El-Ansary, A. Impaired lipid metabolism markers to assess the risk of neuroinflammation in autism spectrum disorder. Metab. Brain Dis. 2018, 33, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Varedi, A.; Rahman, H.; Kumar, D.; Catrow, J.L.; Cox, J.E.; Liu, T.; Florell, S.R.; Boucher, K.M.; Okwundu, N.; Burnett, W.J.; et al. ASA Suppresses PGE2 in Plasma and Melanocytic Nevi of Human Subjects at Increased Risk for Melanoma. Pharmaceuticals 2020, 13, 7. [Google Scholar] [CrossRef]
- Bäck, M.; Hlawaty, H.; Labat, C.; Michel, J.B.; Brink, C. The oral cavity and age: A site of chronic inflammation? PLoS ONE 2007, 2, e1351. [Google Scholar] [CrossRef]
- Ebrahimzadeh, T.; Kuprasertkul, A.; Neugent, M.L.; Lutz, K.C.; Fuentes, J.L.; Gadhvi, J.; Khan, F.; Zhang, C.; Sharon, B.M.; Orth, K.; et al. Urinary prostaglandin E2 as a biomarker for recurrent UTI in postmenopausal women. Life Sci. Alliance 2021, 4, e202000948. [Google Scholar] [CrossRef]
- Doña, I.; Pérez-Sánchez, N.; Eguiluz-Gracia, I.; Muñoz-Cano, R.; Bartra, J.; Torres, M.J.; Cornejo-García, J.A. Progress in understanding hypersensitivity reactions to nonsteroidal anti-inflammatory drugs. Allergy 2020, 75, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Nakamura, T.; Hamasaki, Y.; Yamamoto-Hanada, K.; Fukuie, T.; Narita, M.; Shimosawa, T.; Murata, T.; Ohya, Y. Prostaglandin D(2) metabolite is not a useful clinical indicator for assessing atopic dermatitis. Clin. Exp. Dermatol. 2021, 46, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Nakamura, T.; Takenouchi, S.; Hayashi, A.; Omori, K.; Murata, T. Urinary 8-iso PGF2α and 2,3-dinor-8-iso PGF2α can be indexes of colitis-associated colorectal cancer in mice. PLoS ONE 2021, 16, e0245292. [Google Scholar] [CrossRef]
- Noort, W.A.; van Bulck, B.; Vereecken, A.; de Zwart, F.A.; Keirse, M.J.N.C. Changes in plasma of PGF2α and PGI2 metabolites at and after delivery at term. Prostaglandins 1989, 37, 3–12. [Google Scholar] [CrossRef]
- Martini, D.; Dominguez-Perles, R.; Rosi, A.; Tassotti, M.; Angelino, D.; Medina, S.; Ricci, C.; Guy, A.; Oger, C.; Gigliotti, L.; et al. Effect of Coffee and Cocoa-Based Confectionery Containing Coffee on Markers of DNA Damage and Lipid Peroxidation Products: Results from a Human Intervention Study. Nutrients 2021, 13, 2399. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, R.M.D.; Casciaro, M.M.D.; Rossi, E.M.D.; Calvieri, C.M.D.; Bucci, T.M.D.; Calabrese, C.M.M.D.; Taliani, G.M.D.; Falcone, M.M.D.; Palange, P.M.D.; Bertazzoni, G.M.D.; et al. Platelet Activation Is Associated with Myocardial Infarction in Patients with Pneumonia. J. Am. Coll. Cardiol. 2014, 64, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Song, W.L.; Lawson, J.A.; Wang, M.; Zou, H.; Fitzgerald, G.A. Noninvasive Assessment of the Role of Cyclooxygenases in Cardiovascular Health: A Detailed HPLC/MS/MS Method. Methods Enzymol. 2007, 433, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.M.D.; Pignatelli, P.M.D.; Farcomeni, A.P.; Cangemi, R.M.D.; Hiatt, W.R.M.D.; Bartimoccia, S.P.; Nocella, C.P.; Vicario, T.M.D.; Bucci, T.M.D.; Carnevale, R.P.; et al. Urinary 11-dehydro-thromboxane B2 is associated with cardiovascular events and mortality in patients with atrial fibrillation. Am. Heart J. 2015, 170, 490–497.e1. [Google Scholar] [CrossRef]
- Van der Weiden, R.M.F.; Helmerhorst, F.M.; Keirse, M.J.N.C. Which prostanoid metabolites should be determined for the study of reproductive processes? Prostaglandins Leukot. Essent. Fat. Acids 1998, 58, 205–207. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Prostanoid | Metabolite | Concentration 1 | Comment |
|---|---|---|---|
| PGE2 | Plasma: 3–12 pg/mL | t1/2 < 1min in circulation [27]. | |
| 13,14-dihydro-15-keto PGE2 | Plasma: 10–100 pg/mL [28] | t1/2 = 9 min (in vivo in dogs) [29]. t1/2 = 7 h in protein-free buffer [30]. t1/2 = 3 h in diluted plasma [30]. | |
| tetranor-PGEM | Urine: 7–40 µg per 24 h urine collection (µg/day) [31] | Major urinary metabolite of PGE2 in humans. Marker for systemic PGE2 biosynthesis in vivo. Considered a stable metabolite. | |
| bicyclo PGE2 | Plasma: 20–25 pg/mL [28,32] | Non-enzymatically formed, but base-catalyzed. Considered a stable metabolite. | |
| 13,14-dihydro-15-keto PGA2 | n.d. | Non-enzymatically formed. Considered a stable metabolite. | |
| PGD2 | t1/2 = 0.9 min [33]. | ||
| tetranor-PGDM | Urine: 1.5 ± 0.3 ng/mg creatinine [31] | Major urinary metabolite of PGD2 in humans. Marker for systemic PGD2 biosynthesis in vivo. | |
| 11β-PGF2α | n.d. | ||
| 15d-PGD2 | n.d. | Bioactive metabolite of PGD2; activates PPARγ. | |
| 15-deoxy-Δ12,14-PGJ2 | Plasma: 2 to 350 pg/mL [34] Urine: 6.3 ± 2.7 pg/mg creatinine [35] | ||
| 13,14-dihydro-15-keto PGD2 | n.d. | Not regularly measured as PGD2 metabolite in plasma or urine. | |
| Δ12-PGJ2 | n.d. | Not regularly measured as PGD2 metabolite in plasma or urine. | |
| PGJ2 | n.d. | Not regularly measured as PGD2 metabolite in plasma or urine. | |
| PGF2α | |||
| 13,14-dihydro-15-keto PGF2α | Plasma: 0.08–20 pmol/mL (basal levels) [36]; 40–60 pg/mL (late pregnancy); 1200–4100 pg/mL (parturition) [37] | ||
| tetranor-PGFM | Plasma: 60–100 pg/mL (late pregnancy); 1000 and 2000 pg/mL (parturition); 100–300 pg/mL (24 h after parturition) [37] Urine: 11–59 µg/day (♂), 7–13 µg/day (♀), 2- to 5-fold increase during pregnancy [38] | Major urinary metabolite of PGF2α in humans. | |
| TXA2 | t1/2 = 30 s in circulation [39]. | ||
| TXB2 | 1–2 pg/mL (3–6 fmol/mL) in uninduced blood 300–400 ng/mL (0.8–1.0 nmol/mL) in fully coagulated blood [40] | t1/2 = 7 min [41]. Non-enzymatically formed. Detectable in plasma. Stable metabolite ex vivo. Urinary TXB2 may reflect intrarenal TXA2 production. Serum TXB2 reflects capacity of platelets to synthesize TXA2 in vitro. | |
| 11-dehydro TXB2 | Plasma: 0.9–1.8 pg/mL [42] Urine: 0.9–4.3 pg/mL, 30–70 ng/mmol creatinine, 80–190 pmol/mmol creatinine [43] | Detectable in plasma. t1/2 = 45 min in circulation [42]. Major urinary metabolite of TXA2 in mice. | |
| 2,3-dinor TXB2 | Urine: 10.3 ng/h (138 pg/mg creatine) 45 pmol/mmol creatinine [43] | Urinary 2,3-dinor TXB2 is a marker for systemic TXA2 synthesis in vivo. | |
| PGI2 | t1/2 = 30 s (in vivo). t1/2 = 1.29–1.52 min (in vivo in cats) [44], t1/2 = 6–10 min (in vitro) [45,46,47]. | ||
| 6-keto PGF1α | n.d. | Non-enzymatically formed. Major plasma metabolite of PGI2. Urinary 6-keto PGF1α may reflect intrarenal PGI2 production. | |
| 2,3-dinor-6-keto PGF1α | Urine: 100 pg/mg creatinine [48,49] | Major urinary metabolite of PGI2 in humans. Urinary 2,3-dinor-6-keto PGF1α is a marker for systemic PGI2 synthesis in vivo. |
| Prostanoid Metabolite | Biological Fluid | Association to Disease |
|---|---|---|
| PGE2 | ||
| tetranor-PGEM | Urine | Ulcerative colitis [122]; Viral infections in infants [8]; Breast cancer [123]; Colorectal cancers [124]; Abdominal obesity/pre-diabetes [31]; Cystic fibrosis [125] |
| combined PGE2 and PGE2 metabolites | Plasma/serum/saliva | Obesity [31]; Arterial stiffness [126]; Malaria pathogenesis [127] |
| PGD2 | ||
| tetranor-PGDM | Urine | Cystic fibrosis [125]; Food allergy [128]; Aspirin-intolerant asthma [129]; Astma [130]; Obesity [31]; Duchenne muscular dystrophy [131]; Aspirin-exacerbated respiratory disease [132] |
| 2,3-dinor-11β-PGF2α | Urine | Asthma [130] |
| 13,14-dihydro-15-keto-prostaglandin D2 | Serum | Nonalcoholic fatty liver (NASH) [133] |
| 11β-PGF2α | Serum | Anaphylaxis [134] |
| 15-deoxy-Δ12,14-PGJ2 | Plasma | Diabetes [34] |
| PGF2α | ||
| tetranor-PGFM | Urine | Psoriasis [135] |
| TXA2 | ||
| 2,3-dinor-TXB2 | Urine | Acute coronary syndromes [40] |
| 11-dhydro-TXB2 | Urine | Cardiovascular risk/Acute coronary syndromes [40]; Chronic kidney disease [136]; Cardiovascular disease [137]; Vascular inflammation [138]; Diabetes [139] |
| PGI2 | ||
| 2,3-dinor-6-keto PGF1α | Urine | Diabetes [139]; Acute kidney injury [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idborg, H.; Pawelzik, S.-C. Prostanoid Metabolites as Biomarkers in Human Disease. Metabolites 2022, 12, 721. https://doi.org/10.3390/metabo12080721
Idborg H, Pawelzik S-C. Prostanoid Metabolites as Biomarkers in Human Disease. Metabolites. 2022; 12(8):721. https://doi.org/10.3390/metabo12080721
Chicago/Turabian StyleIdborg, Helena, and Sven-Christian Pawelzik. 2022. "Prostanoid Metabolites as Biomarkers in Human Disease" Metabolites 12, no. 8: 721. https://doi.org/10.3390/metabo12080721
APA StyleIdborg, H., & Pawelzik, S.-C. (2022). Prostanoid Metabolites as Biomarkers in Human Disease. Metabolites, 12(8), 721. https://doi.org/10.3390/metabo12080721