
Toxic Metabolites and Inborn Errors of Amino Acid Metabolism: What One Informs about the Other


Abstract

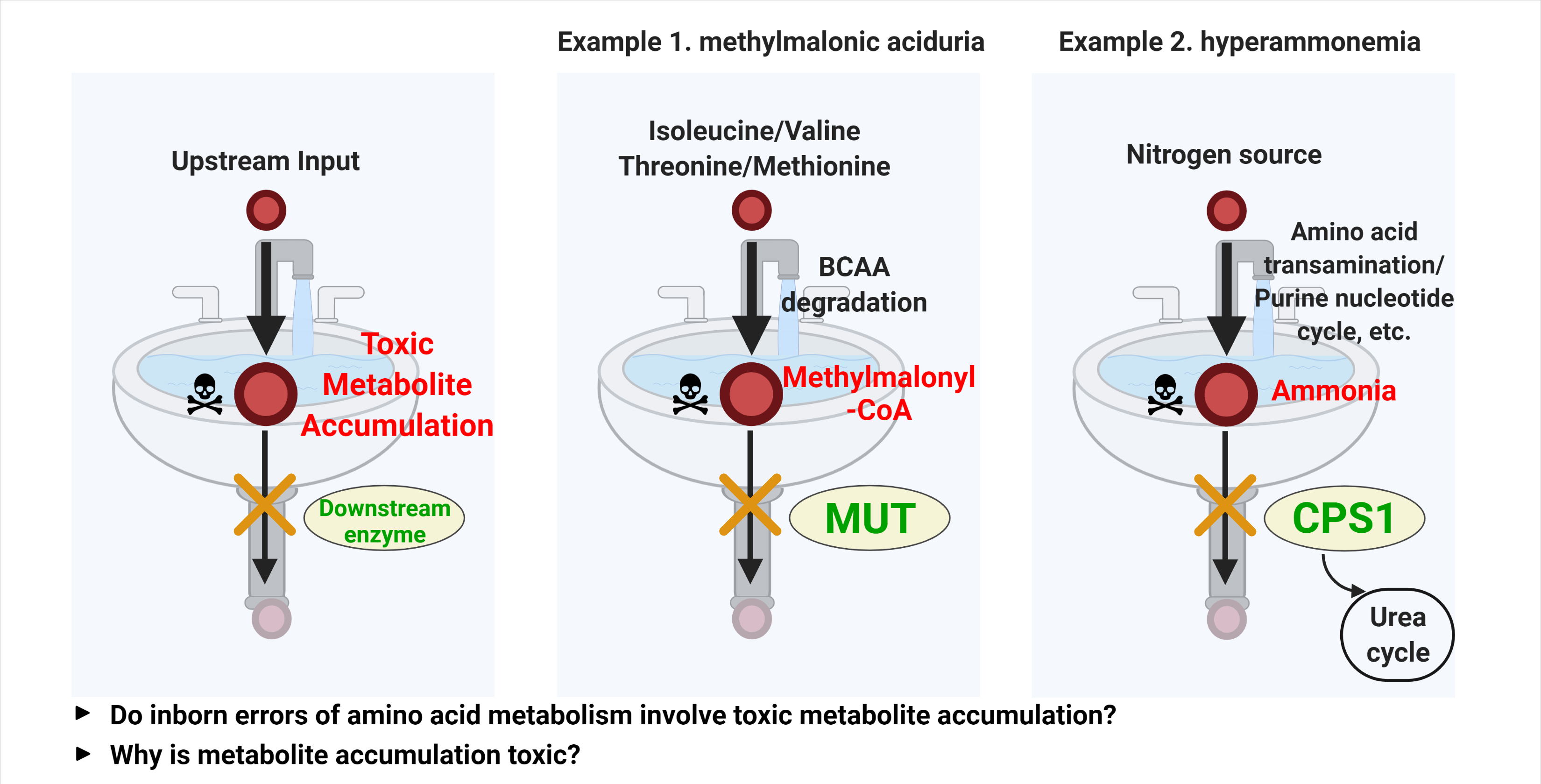
1. Introduction
2. Methylmalonic Acidemia: An IEM and Toxic Metabolite with Broad Implications in Disease
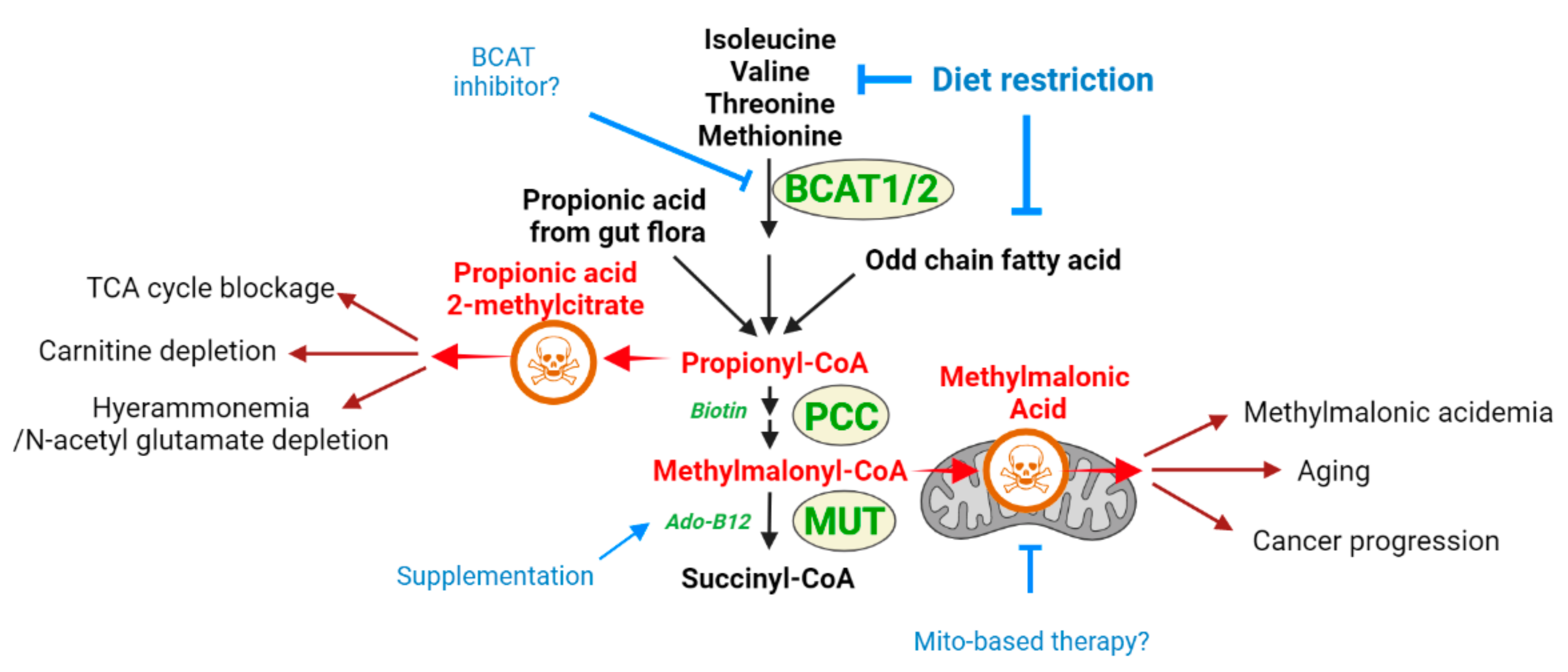
2.1. Methylmalonic Acidemia
2.1.1. Methylmalonic Acid as a Toxic Metabolite Relevant to Multiple Disease States
2.1.2. Methylmalonic Acidemia and MMA as a Toxic Metabolite: What Does One Teach about the Other?
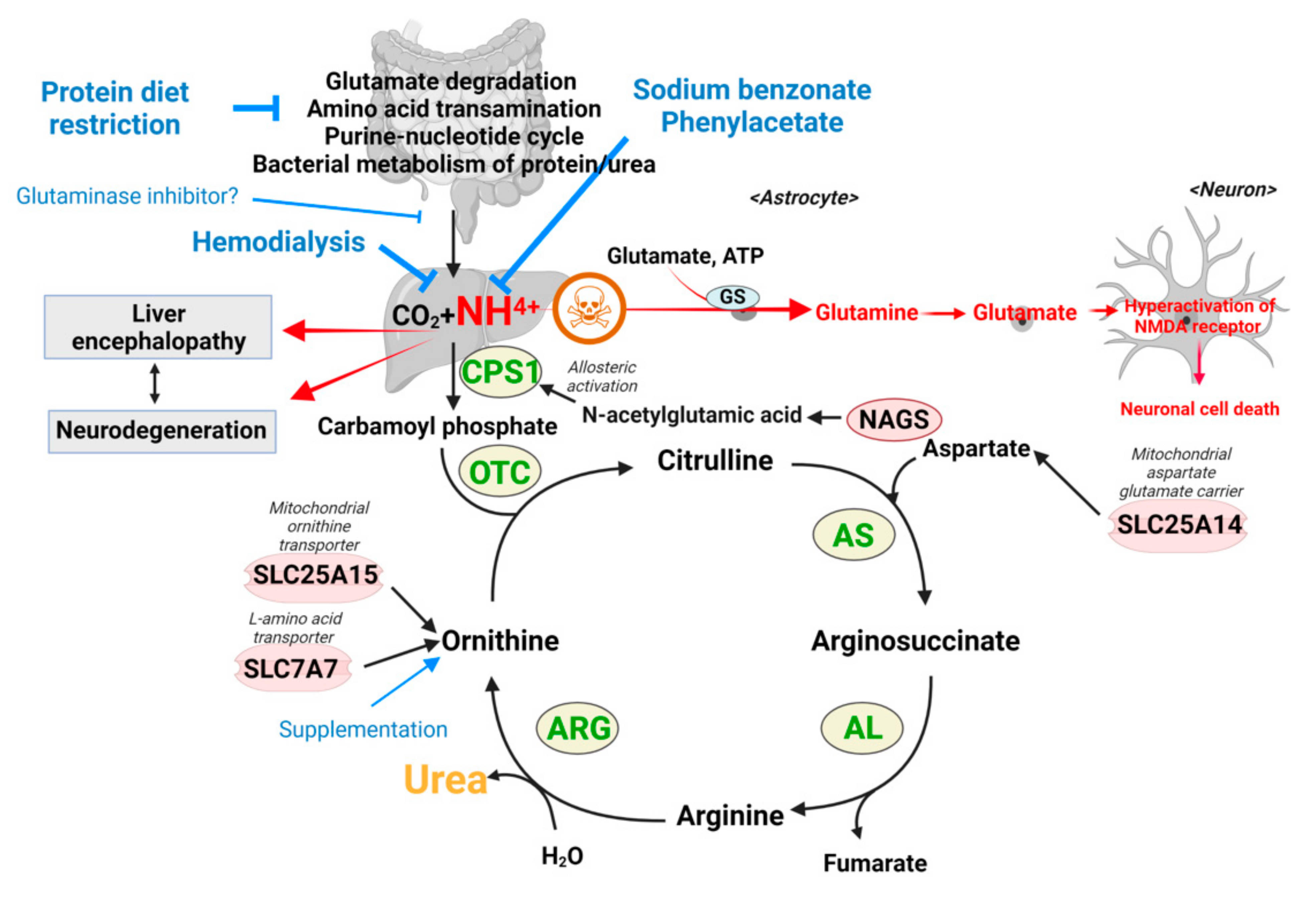
2.2. Hyperammonemia and Ammonia Toxicity: A Second Example
- (i)
- Primary hyperammonemia is caused by the defect of genes involved in the urea cycle. The first and rate-limiting step of the urea cycle is mediated by the carbamoyl phosphate synthetase (CPS), which converts ammonia to carbamoyl phosphate. Ornithine transcarbamylase (OTC) then converts carbamoyl phosphate to citrulline by donating the carbamoyl phosphate group to ornithine. Citrulline is further processed to arginosuccinate by argininosuccinate synthetase (AS) and then to arginine by argininosuccinic acid lyase (AL). As the last step, arginase (ARG) cleaves arginine to produce urea and ornithine, which are excreted or used for another cycle, respectively (Figure 3). Thus, the inactivation of any genes encoding enzymes for the urea cycle leads to perturbation of the urea cycle and accumulation of ammonia. In addition to the five enzymes in the urea cycle, defects of enzymes or transporters supporting the urea cycle can also cause hyperammonemia. For example, a defect of N-acetylglutamate synthase (NAGS), which produces N-acetylglutamic acid (NAcGlu), induces hyperammonemia as CPS activity is allosterically regulated by NAcGlu, a product of NAGS [60]. So, defects in NAGS lead to depletion of NAcGlu, leading to low CPS activity. As another example, hyperammonemia can be induced by defects in relevant transporters, such as mitochondrial ornithine transporter 1 (SLC25A15) and y + L amino acid transporter 1 (SLC7A7) [61,62]. As ornithine is a substrate for OTC enzyme in the urea cycle, the limited availability of ornithine because of defects of ornithine transport can lead to inactivation of the urea cycle. Lastly, the defect of SLC25A14 encoding mitochondrial aspartate glutamate carrier 2 (AGC2), also known as citrin, induces hyperammonemia by limiting the availability of aspartate, the substrate for AS enzyme reaction in the mitochondria [63]. Overall, mutations in any genes that lead to a defective urea cycle can promote primary hyperammonemia.
- (ii)
- Hyperammonemia can also occur as a downstream consequence of other defects, and these cases are categorized as secondary hyperammonemia. In the liver, periportal hepatocytes process a vast amount of ammonia by expressing urea cycle enzymes, and perivenous hepatocytes metabolize ammonia with a low capacity by expressing glutamine synthetase (GS) as a back-up system [64]. Thus, the liver is critical in removing ammonia from the body, and reduced liver function and hepatotoxicity can also lead to hyperammonemia. Secondary hyperammonemia is diagnosed in various pathophysiological contexts, such as acute or chronic liver failure by hepatitis B viral infection, exposure to hepatotoxins, cirrhosis, and urinary tract infection by urease-producing bacteria [56,57]. Another form of secondary hyperammonemia can be observed in patients with propionic acidemia, methylmalonic acidemia, or glutaric acidemia type I. Propionic acidemia is a disease caused by propionyl-CoA carboxylase (PCC) deficiency [65,66]. PCC is an enzyme that processes propionyl-CoA; thus, the mutation in the PCCA or PCCB genes leads to the accumulation of propionyl-CoA. Since propionyl-CoA can inhibit NAGS [67], the urea cycle is slowed, which leads to hyperammonemia. The methylmalonyl-CoA accumulated in the patient with methylmalonic acidemia (see Section 2) also has the same inhibitory activity on NAGS as propionyl-CoA [67], so patients with methylmalonic acidemia showed hyperammonemia as well [67]. Hyperammonemia can be seen in cases and animal models of type I glutaric acidemia [68,69,70]. Thus, a variety of defects or insults can ultimately result in a pathological state of excess ammonia.
2.2.1. Proposed Mechanisms of Ammonia as a Toxic Metabolite
- (1)
- Excessive ammonia perturbs the serotonergic and cholinergic systems. The mouse-bearing mutation in the OTC gene, a hereditary hyperammonemia model, showed significant loss of cholinergic neurons, and the cholinergic neurons were damaged by ammonia in vitro culture [77,78], suggesting a direct toxic effect of ammonia on cholinergic neurons. In terms of underlying mechanisms that may cause toxicity, OTC-mutated mice showed elevated levels of tryptophan, a precursor for serotonin and serotonin metabolite [79], and altered expression of brain serotonin receptors, implying a disturbed serotoninergic system by hyperammonemia [80].
- (2)
- A high level of ammonia disrupts ATP synthesis. The ammonia formed from glutamate deamination can perturb the H+ gradient across the mitochondrial inner membrane due to its alkaline property that binds to H+ [81]. Additionally, the expression and activity of cytochrome C oxidase, the electron transport chain enzyme, are significantly decreased in the hyperammonemia mouse model [82]. Therefore, the H+ gradient required for ATP synthesis is eliminated and thus leads to ATP depletion. Supporting this model, ATP concentration is lower in the brain of the OTC-mutated mouse [83].
- (3)
- The TCA cycle is disrupted by excessive ammonia. The alpha-ketoglutarate dehydrogenase complex (KGDHC), which is required for producing alpha-ketoglutarate and NAD+, is inhibited by the pathological concentration of ammonia [84]. Thus, hyperammonemia induces defects in the TCA cycle.
- (4)
- A high level of ammonia impairs ion homeostasis of astrocytes and neurons. The high level of ammonia perturbs astrocyte potassium buffering by increasing the concentration of extracellular potassium and hyperactivating the Na+-K+-2Cl− cotransporter isoform 1 (NKCC1) in neurons [85]. So, genetic or pharmacological inhibition of NKCC1 has been shown to decrease the clinical and electrophysiological features of ammonia-induced neurotoxicity.
- (5)
- Excessive ammonia impairs cerebral blood flow (CBF), which causes CBF autoregulation failure. Many studies have shown that patients with acute or chronic hyperammonemia suffer from brain edemas due to impaired CBF [86,87,88]. As hyperammonemia interferes with the metabolism of neurotransmitters and ion homeostasis and reduces the metabolic rate, as described above, ammonia accumulation leads to impaired CBF, which is closely regulated by cerebral metabolism.
2.2.2. Ammonia as a Toxic Metabolite Relevant to Other Disease States
2.2.3. What Does One Inform about the Other?
3. Toxic Metabolites Revealed in Inborn Metabolic Disorders May Have Broad Roles in Pathology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Disease | Associated Genes | Accumulated Toxic Metabolites | Toxicity Mechanism | Current Treatment Options |
|---|---|---|---|---|
| Methylmalonic acidemia | MUT | Methylmalonic, hydroxypropionic, methylcitric acids | Energy production deficits via inhibition of TCA cycle and electron transport [36,38,39,40,123] | Restrict branched-chain amino acids Carnitine supplementation Adenosylcobalamin supplementation |
| Primary Hyperammonemia | NAGS, CPS 1, OTC, ASS, SLC25A1, SLC25A14, SLC7A7 | Ammonia | Activation of NMDA receptor by increased glutamate, which is from astrocytes [72] Generation of oxidative stress, inhibition of respiratory chain (energy defects) [74,75,76] | Restrict protein diet Injection of sodium benzoate and phenylacetate Hemodialysis [58] Supplementation of N-carbamoyl-L-glutamic acid (NCG) [105] |
| Phenylketonuria | PAH | Phenylalanine | Restricted transportation of tryptophan and tyrosine at BBB due to high prevalence of phenylalanine occupying L-amino acid transporters [124] Self-assembly of phenylalanine, which is linked to amyloid formation [125] | Restrict protein diet Supplementation of phenylalanine-free amino acid mixture Supplementation of a combination of tyrosine, L-dopa, and 5-hydroxytrytophan [126] Supplementation of tetrahydrobiopterin, which stimulates PAH activity [15] |
| Isovaleric acidemia | IVD | Isocaleryl-CoA derivatives: isovaleric acid, hydroxyisovaleric acid | Inhibition of Na(+), K(+)-ATPase activity via ROS production [127] | Leucin restriction, L-carnitine, and/or glycine supplementation [128,129] |
| Tyrosinemia type 1 | FAH | Fumarylacetoacetate, succinylacetone | Induction of cytochrome c release [117] | Tyrosine restriction Administration of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), the inhibitor for 4-hydroxyphenylpyruvate dioxygenase (HPD), upstream enzyme of fumarylactoacetate [116,130] |
| Maple syrup urine disease | BCKDHA, BCKDHB, DBT | Leucine, isoleucine, and valine | Dysregulated amino and organic acids in brain [131] | Dietary restriction of BCAAs Liver transplantation [131] |
| Glycine encephalopathy | GLDC | Glycine-> Methylglyoxal | Damaged macromolecules (nucleic acids, proteins) as a reactive metabolite [132] | Administration of sodium benzoate to reduce plasma concentration of glycine Blockade of overstimulated NMDA receptors [133] |
| Homocystinuria | CBS MTR | Homocysteine | Induction of oxidative stress, nitrosylation, homocysteinylation, and hypomethylation [134] | Methionine restriction Supplementation with vitamin B6, a cofactor for BHMT that can metabolize homocysteine [135] |
| Propionic academia | PCCA, PCCB | Propionyl-carnitine | Unknown | Restrict protein intake Remove toxic compounds with nitrogen-scavenger medications, extracorporeal detoxification, and/or intravenous carnitine Hemodialysis [48] |
| Cystinuria | SLC3A1, SLC7A9 | Cystine | Formation of cystine stones [136] | Increase fluid intake to increase cystine solubility Administration of thiol drugs [136] |
| 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency | HMGCL | 3-hydroxy-3- methylglutaric acid, 3-methylglutaric acid, 3-hydroxyisovaleric acids | Induction of oxidative stress Disruption of bioenergetics, dynamics, ER-mitochondria communication, and signaling pathways [137] Induction of DNA damage [138] | Restrict leucine diet Supplementary glucose [139,140] |
| Hyperprolinemia | PRODH ALDH4A1 | Proline | Excitotoxin [141] Reduction in glutamate uptake, Na(+), K(+)-ATPase activity, ATP levels Increased lipid peroxidation [142] | Restriction of dietary proline [143] |
| Hyperlysinemia | AASS | Lysine, Pipecolic acid | Unknown | Dietary restriction of lysine [144] |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Romero, P.; Wagg, J.; Green, M.L.; Kaiser, D.; Krummenacker, M.; Karp, P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2005, 6, R2. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W. Inborn errors of metabolism. Clin. Perinatol. 2015, 42, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; van Karnebeek, C.D.M. Inborn errors of metabolism. Handb. Clin. Neurol. 2019, 162, 449–481. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.M.; Garcia-Cazorla, A. Inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatric Clin. N. Am. 2018, 65, 179–208. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, E.; Mienie, L.J.; de Vries, W.N.; de Wet, W.J.; Carlsson, B.; Larsson, A. Prenatal analysis in two suspected cases of glutathione synthetase deficiency. J. Inherit. Metab. Dis. 1993, 16, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Zetterstrom, R.; Hagenfeldt, L.; Andersson, R.; Dreborg, S.; Hornell, H. Pyroglutamic aciduria (5-oxoprolinuria), an inborn error in glutathione metabolism. Pediatric Res. 1974, 8, 852–856. [Google Scholar] [CrossRef][Green Version]
- Wellner, V.P.; Sekura, R.; Meister, A.; Larsson, A. Glutathione synthetase deficiency, an inborn error of metabolism involving the gamma-glutamyl cycle in patients with 5-oxoprolinuria (pyroglutamic aciduria). Proc. Natl. Acad. Sci. USA 1974, 71, 2505–2509. [Google Scholar] [CrossRef]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007, 2, 16. [Google Scholar] [CrossRef]
- Ristoff, E.; Mayatepek, E.; Larsson, A. Long-term clinical outcome in patients with glutathione synthetase deficiency. J. Pediatric 2001, 139, 79–84. [Google Scholar] [CrossRef]
- Stockler, S.; Holzbach, U.; Hanefeld, F.; Marquardt, I.; Helms, G.; Requart, M.; Hanicke, W.; Frahm, J. Creatine deficiency in the brain: A new, treatable inborn error of metabolism. Pediatric Res. 1994, 36, 409–413. [Google Scholar] [CrossRef]
- Stockler, S.; Isbrandt, D.; Hanefeld, F.; Schmidt, B.; von Figura, K. Guanidinoacetate methyltransferase deficiency: The first inborn error of creatine metabolism in man. Am. J. Hum. Genet. 1996, 58, 914–922. [Google Scholar] [PubMed]
- Mainka, T.; Fischer, J.F.; Huebl, J.; Jung, A.; Lier, D.; Mosejova, A.; Skorvanek, M.; de Koning, T.J.; Kuhn, A.A.; Freisinger, P.; et al. The neurological and neuropsychiatric spectrum of adults with late-treated phenylketonuria. Parkinsonism Relat. Disord. 2021, 89, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.J. Neurological deterioration in young adults with phenylketonuria. Lancet 1990, 336, 949. [Google Scholar] [CrossRef][Green Version]
- Smith, I.; Clayton, B.E.; Wolff, O.H. New variant of phenylketonuria with progressive neurological illness unresponsive to phenylalanine restriction. Lancet 1975, 1, 1108–1111. [Google Scholar] [CrossRef]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Blau, N. Genetics of Phenylketonuria: Then and Now. Hum. Mutat. 2016, 37, 508–515. [Google Scholar] [CrossRef]
- Bayat, A.; Moller, L.B.; Lund, A.M. Diagnostics and treatment of phenylketonuria. Ugeskr. Laeger 2015, 177, V07140383. [Google Scholar]
- Berry, S.A. Newborn screening. Clin. Perinatol. 2015, 42, 441–453. [Google Scholar] [CrossRef]
- Ledley, F.D. Perspectives on methylmalonic acidemia resulting from molecular cloning of methylmalonyl CoA mutase. Bioessays 1990, 12, 335–340. [Google Scholar] [CrossRef]
- Ledley, F.D.; Rosenblatt, D.S. Mutations in mut methylmalonic acidemia: Clinical and enzymatic correlations. Hum. Mutat. 1997, 9, 1–6. [Google Scholar] [CrossRef]
- Zhou, X.; Cui, Y.; Han, J. Methylmalonic acidemia: Current status and research priorities. Intractable Rare Dis. Res. 2018, 7, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Caterino, M.; Cevenini, A.; Jung, V.; Chhuon, C.; Lipecka, J.; Fedele, R.; Guerrera, I.C.; Ruoppolo, M. Proteomics Reveals that Methylmalonyl-CoA Mutase Modulates Cell Architecture and Increases Susceptibility to Stress. Int. J. Mol. Sci. 2020, 21, 4998. [Google Scholar] [CrossRef] [PubMed]
- Barash, V.; Elpeleg, O.; Amit, R.; Gottfried, S.; Yatziv, S.; Gutman, A. Propionic acidemia--biochemical studies. Isr. J. Med. Sci. 1989, 25, 103–106. [Google Scholar] [PubMed]
- Manoli, I.; Sloan, J.L.; Venditti, C.P. Isolated Methylmalonic Acidemia. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Baumgartner, M.R.; Horster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef]
- Kolker, S.; Okun, J.G. Methylmalonic acid—An endogenous toxin? Cell. Mol. Life Sci. 2005, 62, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.D.; Elmaghrabi, A.; Gotway, G.; Wolf, M.T.F. The roles of homocysteinemia and methylmalonic acidemia in kidney injury in atypical hemolytic uremic syndrome caused by cobalamin C deficiency. Pediatric Nephrol. 2021, 37, 1415–1418. [Google Scholar] [CrossRef]
- Brox-Torrecilla, N.; Arhip, L.; Miguelez-Gonzalez, M.; Castellano-Gasch, S.; Contreras-Chicote, A.; Rodriguez-Ferrero, M.L.; Motilla de la Camara, M.L.; Serrano-Moreno, C.; Cuerda Compes, C. Late-onset methylmalonic acidemia and homocysteinemia. Nutr. Hosp. 2021, 38, 871–875. [Google Scholar] [CrossRef]
- Chen, T.; Liang, L.; Zhang, H.; Ye, J.; Qiu, W.; Xiao, B.; Zhu, H.; Wang, L.; Xu, F.; Gong, Z.; et al. Value of amniotic fluid homocysteine assay in prenatal diagnosis of combined methylmalonic acidemia and homocystinuria, cobalamin C type. Orphanet J. Rare Dis. 2021, 16, 125. [Google Scholar] [CrossRef]
- Morris, M.S.; Jacques, P.F.; Rosenberg, I.H.; Selhub, J. Elevated serum methylmalonic acid concentrations are common among elderly Americans. J. Nutr. 2002, 132, 2799–2803. [Google Scholar] [CrossRef]
- Gomes, A.P.; Ilter, D.; Low, V.; Endress, J.E.; Fernandez-Garcia, J.; Rosenzweig, A.; Schild, T.; Broekaert, D.; Ahmed, A.; Planque, M.; et al. Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature 2020, 585, 283–287. [Google Scholar] [CrossRef]
- Wolters, M.; Strohle, A.; Hahn, A. Age-associated changes in the metabolism of vitamin B(12) and folic acid: Prevalence, aetiopathogenesis and pathophysiological consequences. Z. Gerontol. Geriatr. 2004, 37, 109–135. [Google Scholar] [CrossRef]
- Wong, C.W. Vitamin B12 deficiency in the elderly: Is it worth screening? Hong Kong Med. J. 2015, 21, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Oh, R.; Brown, D.L. Vitamin B12 deficiency. Am. Fam. Physician 2003, 67, 979–986. [Google Scholar]
- Nilsson-Ehle, H. Age-related changes in cobalamin (vitamin B12) handling. Implications for therapy. Drugs Aging 1998, 12, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Okun, J.G.; Horster, F.; Farkas, L.M.; Feyh, P.; Hinz, A.; Sauer, S.; Hoffmann, G.F.; Unsicker, K.; Mayatepek, E.; Kolker, S. Neurodegeneration in methylmalonic aciduria involves inhibition of complex II and the tricarboxylic acid cycle, and synergistically acting excitotoxicity. J. Biol. Chem. 2002, 277, 14674–14680. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.C.; Turton, N.; Boan, E.J.; Bennett, E.; Philips, S.; Heaton, R.A.; Hargreaves, I.P. The Effect of Methylmalonic Acid Treatment on Human Neuronal Cell Coenzyme Q10 Status and Mitochondrial Function. Int. J. Mol. Sci. 2020, 21, 9137. [Google Scholar] [CrossRef]
- Dutra, J.C.; Dutra-Filho, C.S.; Cardozo, S.E.; Wannmacher, C.M.; Sarkis, J.J.; Wajner, M. Inhibition of succinate dehydrogenase and beta-hydroxybutyrate dehydrogenase activities by methylmalonate in brain and liver of developing rats. J. Inherit. Metab. Dis. 1993, 16, 147–153. [Google Scholar] [CrossRef]
- Brusque, A.M.; Borba Rosa, R.; Schuck, P.F.; Dalcin, K.B.; Ribeiro, C.A.; Silva, C.G.; Wannmacher, C.M.; Dutra-Filho, C.S.; Wyse, A.T.; Briones, P.; et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochem. Int. 2002, 40, 593–601. [Google Scholar] [CrossRef]
- Toyoshima, S.; Watanabe, F.; Saido, H.; Miyatake, K.; Nakano, Y. Methylmalonic acid inhibits respiration in rat liver mitochondria. J. Nutr. 1995, 125, 2846–2850. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, D.; Wang, K.; Kwok, T. High Serum Folate Is Associated with Brain Atrophy in Older Diabetic People with Vitamin B12 Deficiency. J. Nutr. Health Aging 2017, 21, 1065–1071. [Google Scholar] [CrossRef]
- Allen, R.H.; Stabler, S.P.; Lindenbaum, J. Relevance of vitamins, homocysteine and other metabolites in neuropsychiatric disorders. Eur. J. Pediatric 1998, 157 (Suppl. S2), S122–S126. [Google Scholar] [CrossRef] [PubMed]
- Forny, P.; Hochuli, M.; Rahman, Y.; Deheragoda, M.; Weber, A.; Baruteau, J.; Grunewald, S. Liver neoplasms in methylmalonic aciduria: An emerging complication. J. Inherit. Metab. Dis. 2019, 42, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.; Mascarenhas, L.; Boles, R.G.; Kerkar, N.; Genyk, Y.; Venkatramani, R. Hepatoblastoma in a patient with methylmalonic aciduria. Am. J. Med. Genet. A 2015, 167A, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Cosson, M.A.; Touati, G.; Lacaille, F.; Valayannnopoulos, V.; Guyot, C.; Guest, G.; Verkarre, V.; Chretien, D.; Rabier, D.; Munnich, A.; et al. Liver hepatoblastoma and multiple OXPHOS deficiency in the follow-up of a patient with methylmalonic aciduria. Mol. Genet. Metab. 2008, 95, 107–109. [Google Scholar] [CrossRef]
- Hauser, N.S.; Manoli, I.; Graf, J.C.; Sloan, J.; Venditti, C.P. Variable dietary management of methylmalonic acidemia: Metabolic and energetic correlations. Am. J. Clin. Nutr. 2011, 93, 47–56. [Google Scholar] [CrossRef]
- Ney, D.; Bay, C.; Saudubray, J.M.; Kelts, D.G.; Kulovich, S.; Sweetman, L.; Nyhan, W.L. An evaluation of protein requirements in methylmalonic acidaemia. J. Inherit. Metab. Dis. 1985, 8, 132–142. [Google Scholar] [CrossRef]
- Shchelochkov, O.A.; Carrillo, N.; Venditti, C. Propionic Acidemia. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hu, L.Y.; Boxer, P.A.; Kesten, S.R.; Lei, H.J.; Wustrow, D.J.; Moreland, D.W.; Zhang, L.; Ahn, K.; Ryder, T.R.; Liu, X.; et al. The design and synthesis of human branched-chain amino acid aminotransferase inhibitors for treatment of neurodegenerative diseases. Bioorg. Med. Chem. Lett. 2006, 16, 2337–2340. [Google Scholar] [CrossRef]
- Scanlon, M. Inhibitors of BCATm: A Tough Nut To Crack. J. Med. Chem. 2015, 58, 7138–7139. [Google Scholar] [CrossRef]
- Papathanassiu, A.E.; Ko, J.H.; Imprialou, M.; Bagnati, M.; Srivastava, P.K.; Vu, H.A.; Cucchi, D.; McAdoo, S.P.; Ananieva, E.A.; Mauro, C.; et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat. Commun. 2017, 8, 16040. [Google Scholar] [CrossRef]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. EBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef]
- Stepien, K.M.; Heaton, R.; Rankin, S.; Murphy, A.; Bentley, J.; Sexton, D.; Hargreaves, I.P. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J. Clin. Med. 2017, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.G.; Borges, C.G.; Seminotti, B.; Amaral, A.U.; Knebel, L.A.; Eichler, P.; de Oliveira, A.B.; Leipnitz, G.; Wajner, M. Experimental evidence that methylmalonic acid provokes oxidative damage and compromises antioxidant defenses in nerve terminal and striatum of young rats. Cell. Mol. Neurobiol. 2011, 31, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Doets, E.L.; van Wijngaarden, J.P.; Szczecinska, A.; Dullemeijer, C.; Souverein, O.W.; Dhonukshe-Rutten, R.A.; Cavelaars, A.E.; van ‘t Veer, P.; Brzozowska, A.; de Groot, L.C. Vitamin B12 intake and status and cognitive function in elderly people. Epidemiol. Rev. 2013, 35, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Auron, A.; Brophy, P.D. Hyperammonemia in review: Pathophysiology, diagnosis, and treatment. Pediatric Nephrol. 2012, 27, 207–222. [Google Scholar] [CrossRef]
- Leger, R.F.; Silverman, M.S.; Hauck, E.S.; Guvakova, K.D. Hyperammonemia Post Lung Transplantation: A Review. Clin. Med. Insights Circ. Respir. Pulm. Med. 2020, 14, 1179548420966234. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Nagalli, S. Hyperammonemia; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Haberle, J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch. Biochem. Biophys. 2013, 536, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Mian, A.; Lee, B. Urea-cycle disorders as a paradigm for inborn errors of hepatocyte metabolism. Trends Mol. Med. 2002, 8, 583–589. [Google Scholar] [CrossRef]
- Nunes, V.; Niinikoski, H. Lysinuric Protein Intolerance. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Noguchi, A.; Takahashi, T. Overview of symptoms and treatment for lysinuric protein intolerance. J. Hum. Genet. 2019, 64, 849–858. [Google Scholar] [CrossRef]
- Saheki, T.; Inoue, K.; Tushima, A.; Mutoh, K.; Kobayashi, K. Citrin deficiency and current treatment concepts. Mol. Genet. Metab. 2010, 100 (Suppl S1), S59–S64. [Google Scholar] [CrossRef]
- Haberle, J. Clinical practice: The management of hyperammonemia. Eur. J. Pediatric 2011, 170, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Haberle, J.; Chakrapani, A.; Ah Mew, N.; Longo, N. Hyperammonaemia in classic organic acidaemias: A review of the literature and two case histories. Orphanet J. Rare Dis. 2018, 13, 219. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, H.R.; Ernst, S.L.; Ashurst, C.L.; Pasquali, M.; Longo, N. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Mol. Genet. Metab. 2006, 88, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Dercksen, M.; IJlst, L.; Duran, M.; Mienie, L.J.; van Cruchten, A.; van der Westhuizen, F.H.; Wanders, R.J. Inhibition of N-acetylglutamate synthase by various monocarboxylic and dicarboxylic short-chain coenzyme A esters and the production of alternative glutamate esters. Biochim. Biophys. Acta 2014, 1842, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Melo, M.; Remacle, N.; Cudre-Cung, H.P.; Roux, C.; Poms, M.; Cudalbu, C.; Barroso, M.; Gersting, S.W.; Feichtinger, R.G.; Mayr, J.A.; et al. The first knock-in rat model for glutaric aciduria type I allows further insights into pathophysiology in brain and periphery. Mol. Genet. Metab. 2021, 133, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Melo, M.; Fontana, A.O.; Viertl, D.; Allenbach, G.; Prior, J.O.; Rotman, S.; Feichtinger, R.G.; Mayr, J.A.; Costanzo, M.; Caterino, M.; et al. A knock-in rat model unravels acute and chronic renal toxicity in glutaric aciduria type I. Mol. Genet. Metab. 2021, 134, 287–300. [Google Scholar] [CrossRef]
- Hafeez, A.; Fatima, S.; Chaudhry, N.; Khadim, M.T. Dyskinesia in a Child: A Concern for a Rare Neuro-Metabolic Disorder. J. Coll. Physicians Surg. Pak. 2019, 29, 84–86. [Google Scholar] [CrossRef]
- Braissant, O.; McLin, V.A.; Cudalbu, C. Ammonia toxicity to the brain. J. Inherit. Metab. Dis. 2013, 36, 595–612. [Google Scholar] [CrossRef]
- Cooper, A.J. Role of glutamine in cerebral nitrogen metabolism and ammonia neurotoxicity. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef]
- Llansola, M.; Rodrigo, R.; Monfort, P.; Montoliu, C.; Kosenko, E.; Cauli, O.; Piedrafita, B.; El Mlili, N.; Felipo, V. NMDA receptors in hyperammonemia and hepatic encephalopathy. Metab. Brain Dis. 2007, 22, 321–335. [Google Scholar] [CrossRef]
- Klejman, A.; Wegrzynowicz, M.; Szatmari, E.M.; Mioduszewska, B.; Hetman, M.; Albrecht, J. Mechanisms of ammonia-induced cell death in rat cortical neurons: Roles of NMDA receptors and glutathione. Neurochem. Int. 2005, 47, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, R.V.; da Silva Ferreira, F.; Heimfarth, L.; Pierozan, P.; Fernandes, C.; Pessoa-Pureur, R. Acute Hyperammonemia Induces NMDA-Mediated Hypophosphorylation of Intermediate Filaments Through PP1 and PP2B in Cerebral Cortex of Young Rats. Neurotox. Res. 2016, 30, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Ratnakumari, L.; Qureshi, I.A.; Butterworth, R.F. Evidence for cholinergic neuronal loss in brain in congenital ornithine transcarbamylase deficiency. Neurosci. Lett. 1994, 178, 63–65. [Google Scholar] [CrossRef]
- Braissant, O.; Henry, H.; Villard, A.M.; Zurich, M.G.; Loup, M.; Eilers, B.; Parlascino, G.; Matter, E.; Boulat, O.; Honegger, P.; et al. Ammonium-induced impairment of axonal growth is prevented through glial creatine. J. Neurosci. 2002, 22, 9810–9820. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.; Colombo, J.P. Increase of tryptophan and 5-hydroxyindole acetic acid in the brain of ornithine carbamoyltransferase deficient sparse-fur mice. Pediatric Res. 1984, 18, 372–375. [Google Scholar] [CrossRef]
- Robinson, M.B.; Anegawa, N.J.; Gorry, E.; Qureshi, I.A.; Coyle, J.T.; Lucki, I.; Batshaw, M.L. Brain serotonin2 and serotonin1A receptors are altered in the congenitally hyperammonemic sparse fur mouse. J. Neurochem. 1992, 58, 1016–1022. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef]
- Rao, K.V.; Mawal, Y.R.; Qureshi, I.A. Progressive decrease of cerebral cytochrome C oxidase activity in sparse-fur mice: Role of acetyl-L-carnitine in restoring the ammonia-induced cerebral energy depletion. Neurosci. Lett. 1997, 224, 83–86. [Google Scholar] [CrossRef]
- Ratnakumari, L.; Qureshi, I.A.; Butterworth, R.F. Effects of congenital hyperammonemia on the cerebral and hepatic levels of the intermediates of energy metabolism in spf mice. Biochem. Biophys. Res. Commun. 1992, 184, 746–751. [Google Scholar] [CrossRef]
- Lai, J.C.; Cooper, A.J. Brain alpha-ketoglutarate dehydrogenase complex: Kinetic properties, regional distribution, and effects of inhibitors. J. Neurochem. 1986, 47, 1376–1386. [Google Scholar] [CrossRef]
- Rangroo Thrane, V.; Thrane, A.S.; Wang, F.; Cotrina, M.L.; Smith, N.A.; Chen, M.; Xu, Q.; Kang, N.; Fujita, T.; Nagelhus, E.A.; et al. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 2013, 19, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Toftengi, F.; Larsen, F.S. Management of patients with fulminant hepatic failure and brain edema. Metab. Brain Dis. 2004, 19, 207–214. [Google Scholar] [CrossRef]
- Bjerring, P.N.; Eefsen, M.; Hansen, B.A.; Larsen, F.S. The brain in acute liver failure. A tortuous path from hyperammonemia to cerebral edema. Metab. Brain Dis. 2009, 24, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.S. Cerebral blood flow in hyperammonemia: Heterogeneity and starling forces in capillaries. Metab. Brain Dis. 2002, 17, 229–235. [Google Scholar] [CrossRef]
- Fisman, M.; Gordon, B.; Feleki, V.; Helmes, E.; Appell, J.; Rabheru, K. Hyperammonemia in Alzheimer’s disease. Am. J. Psychiatry 1985, 142, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Patten, B.M.; Kurlander, H.M.; Evans, B. Free amino acid concentrations in spinal tissue from patients dying of motor neuron disease. Acta Neurol. Scand. 1982, 66, 594–599. [Google Scholar] [CrossRef]
- Jo, D.; Kim, B.C.; Cho, K.A.; Song, J. The Cerebral Effect of Ammonia in Brain Aging: Blood-Brain Barrier Breakdown, Mitochondrial Dysfunction, and Neuroinflammation. J. Clin. Med. 2021, 10, 2773. [Google Scholar] [CrossRef]
- Eiser, A.R.; Fulop, T. Extra-cranial factors in the development of Alzheimer’s disease. Brain Res. 2020, 1748, 147076. [Google Scholar] [CrossRef]
- Aguilar, M.A.; Minarro, J.; Felipo, V. Chronic moderate hyperammonemia impairs active and passive avoidance behavior and conditional discrimination learning in rats. Exp. Neurol. 2000, 161, 704–713. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Sujatha, R.; Paul, V. Effect of ammonia on motor function in adult rats. Brain Res. Bull. 1997, 43, 275–278. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Norenberg, M.D. Glutamine Synthetase: Role in Neurological Disorders. Adv. Neurobiol. 2016, 13, 327–350. [Google Scholar] [CrossRef] [PubMed]
- Owen, E.E.; Johnson, J.H.; Tyor, M.P. The effect of induced hyperammonemia on renal ammonia metabolism. J. Clin. Investig. 1961, 40, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Bento, L.M.; Carvalheira, J.B.; Menegon, L.F.; Saad, M.J.; Gontijo, J.A. Effects of NH4Cl intake on renal growth in rats: Role of MAPK signalling pathway. Nephrol. Dial. Transplant. 2005, 20, 2654–2660. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Panickar, K.S.; Jayakumar, A.R.; Rao, K.V.; Norenberg, M.D. Ammonia-induced activation of p53 in cultured astrocytes: Role in cell swelling and glutamate uptake. Neurochem. Int. 2009, 55, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Jalan, R.; De Chiara, F.; Balasubramaniyan, V.; Andreola, F.; Khetan, V.; Malago, M.; Pinzani, M.; Mookerjee, R.P.; Rombouts, K. Ammonia produces pathological changes in human hepatic stellate cells and is a target for therapy of portal hypertension. J. Hepatol. 2016, 64, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Mittal, G.; Sultana, S.; Bhatnagar, A. Ameliorative potential of alpha-ketoglutaric acid (AKG) on acute lung injuries induced by ammonia inhalation in rats. Exp. Lung Res. 2012, 38, 435–444. [Google Scholar] [CrossRef]
- Mirbod, F.; Schaller, R.A.; Cole, G.T. Purification and characterization of urease isolated from the pathogenic fungus Coccidioides immitis. Med. Mycol. 2002, 40, 35–44. [Google Scholar] [CrossRef][Green Version]
- Wise, H.Z.; Hung, C.Y.; Whiston, E.; Taylor, J.W.; Cole, G.T. Extracellular ammonia at sites of pulmonary infection with Coccidioides posadasii contributes to severity of the respiratory disease. Microb. Pathog. 2013, 59–60, 19–28. [Google Scholar] [CrossRef]
- Lichtenstein, G.R.; Kaiser, L.R.; Tuchman, M.; Palevsky, H.I.; Kotloff, R.M.; O’Brien, C.B.; Furth, E.E.; Raps, E.C.; Berry, G.T. Fatal hyperammonemia following orthotopic lung transplantation. Gastroenterology 1997, 112, 236–240. [Google Scholar] [CrossRef]
- Camacho, J.; Rioseco-Camacho, N. Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chapel-Crespo, C.C.; Diaz, G.A.; Oishi, K. Efficacy of N-carbamoyl-L-glutamic acid for the treatment of inherited metabolic disorders. Expert Rev. Endocrinol. Metab. 2016, 11, 467–473. [Google Scholar] [CrossRef]
- Haberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- De Las Heras, J.; Aldamiz-Echevarria, L.; Martinez-Chantar, M.L.; Delgado, T.C. An update on the use of benzoate, phenylacetate and phenylbutyrate ammonia scavengers for interrogating and modifying liver nitrogen metabolism and its implications in urea cycle disorders and liver disease. Expert Opin. Drug Metab. Toxicol. 2017, 13, 439–448. [Google Scholar] [CrossRef]
- Khasnavis, S.; Pahan, K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neuroimmune Pharmacol. 2014, 9, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Khasnavis, S.; Pahan, K. Sodium benzoate, a metabolite of cinnamon and a food additive, upregulates neuroprotective Parkinson disease protein DJ-1 in astrocytes and neurons. J. Neuroimmune Pharmacol. 2012, 7, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Modi, K.K.; Roy, A.; Brahmachari, S.; Rangasamy, S.B.; Pahan, K. Cinnamon and Its Metabolite Sodium Benzoate Attenuate the Activation of p21rac and Protect Memory and Learning in an Animal Model of Alzheimer’s Disease. PLoS ONE 2015, 10, e0130398. [Google Scholar] [CrossRef] [PubMed]
- Modi, K.K.; Rangasamy, S.B.; Dasarathi, S.; Roy, A.; Pahan, K. Cinnamon Converts Poor Learning Mice to Good Learners: Implications for Memory Improvement. J. Neuroimmune Pharmacol. 2016, 11, 693–707. [Google Scholar] [CrossRef]
- Ahmed, H.; Haider, A.; Ametamey, S.M. N-Methyl-D-Aspartate (NMDA) receptor modulators: A patent review (2015–present). Expert Opin. Ther. Pat. 2020, 30, 743–767. [Google Scholar] [CrossRef]
- Diaz-Herrero, M.M.; del Campo, J.A.; Carbonero-Aguilar, P.; Vega-Pérez, J.M.; Iglesias-Guerra, F.; Periñán, I.; Miñano, F.J.; Bautista, J.; Romero-Gómez, M. THDP17 Decreases Ammonia Production through Glutaminase Inhibition. A New Drug for Hepatic Encephalopathy Therapy. PLoS ONE 2014, 9, e109787. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Rama Rao, K.V.; Jayakumar, A.R. Ammonia Neurotoxicity and the Mitochondrial Permeability Transition. J. Bioenerg. Biomembr. 2004, 36, 303–307. [Google Scholar] [CrossRef]
- Russo, P.A.; Mitchell, G.A.; Tanguay, R.M. Tyrosinemia: A review. Pediatric Dev. Pathol. 2001, 4, 212–221. [Google Scholar] [CrossRef]
- Holme, E.; Lindstedt, S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J. Inherit. Metab. Dis. 1998, 21, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Sun, M.; Miyahara, M.; Umeyama, K.; Urakami, K.; Yamamoto, T.; Jakobs, C.; Matsuda, I.; Endo, F. Hepatocyte injury in tyrosinemia type 1 is induced by fumarylacetoacetate and is inhibited by caspase inhibitors. Proc. Natl. Acad. Sci. USA 1998, 95, 9552–9557. [Google Scholar] [CrossRef] [PubMed]
- Benevenga, N.J.; Steele, R.D. Adverse effects of excessive consumption of amino acids. Annu. Rev. Nutr. 1984, 4, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Nino, M.D. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients 2017, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Bjoraker, K.J.; Swanson, M.A.; Coughlin, C.R., 2nd; Christodoulou, J.; Tan, E.S.; Fergeson, M.; Dyack, S.; Ahmad, A.; Friederich, M.W.; Spector, E.B.; et al. Neurodevelopmental Outcome and Treatment Efficacy of Benzoate and Dextromethorphan in Siblings with Attenuated Nonketotic Hyperglycinemia. J. Pediatric 2016, 170, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, J.L.; Vande Kerckhove, K.; Hennermann, J.B.; Mahieu, V.; Declercq, P.; Mertens, S.; De Becker, M.; Kishnani, P.S.; Jaeken, J. Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J. Inherit. Metab. Dis. 2005, 28, 651–663. [Google Scholar] [CrossRef]
- Wiltshire, E.J.; Poplawski, N.K.; Harrison, J.R.; Fletcher, J.M. Treatment of late-onset nonketotic hyperglycinaemia: Effectiveness of imipramine and benzoate. J. Inherit. Metab. Dis. 2000, 23, 15–21. [Google Scholar] [CrossRef]
- Wajner, M.; Coelho, J.C. Neurological dysfunction in methylmalonic acidaemia is probably related to the inhibitory effect of methylmalonate on brain energy production. J. Inherit. Metab. Dis. 1997, 20, 761–768. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Hoeksma, M.; Reijngoud, D.J. Brain dysfunction in phenylketonuria: Is phenylalanine toxicity the only possible cause? J. Inherit. Metab. Dis. 2009, 32, 46–51. [Google Scholar] [CrossRef]
- Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno, A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. [Google Scholar] [CrossRef]
- Bonafe, L.; Blau, N.; Burlina, A.P.; Romstad, A.; Guttler, F.; Burlina, A.B. Treatable neurotransmitter deficiency in mild phenylketonuria. Neurology 2001, 57, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.A.; Balestro, F.; Grando, V.; Wajner, M. Isovaleric acid reduces Na+, K+-ATPase activity in synaptic membranes from cerebral cortex of young rats. Cell. Mol. Neurobiol. 2007, 27, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Chinen, Y.; Nakamura, S.; Tamashiro, K.; Sakamoto, O.; Tashiro, K.; Inokuchi, T.; Nakanishi, K. Isovaleric acidemia: Therapeutic response to supplementation with glycine, l-carnitine, or both in combination and a 10-year follow-up case study. Mol. Genet. Metab. Rep. 2017, 11, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Fries, M.H.; Rinaldo, P.; Schmidt-Sommerfeld, E.; Jurecki, E.; Packman, S. Isovaleric acidemia: Response to a leucine load after three weeks of supplementation with glycine, L-carnitine, and combined glycine-carnitine therapy. J. Pediatric 1996, 129, 449–452. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Q.; Yang, H.; Zou, Q.; Lai, C.; Jiang, F.; Zhao, P.; Luo, Z.; Yang, J.; Chen, Q.; et al. Fumarylacetoacetate Hydrolase Knock-out Rabbit Model for Hereditary Tyrosinemia Type 1. J. Biol. Chem. 2017, 292, 4755–4763. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jakher, Y.; Ahrens-Nicklas, R.C. Brain Branched-Chain Amino Acids in Maple Syrup Urine Disease: Implications for Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 7490. [Google Scholar] [CrossRef]
- Kalapos, M.P. Methylglyoxal toxicity in mammals. Toxicol. Lett. 1994, 73, 3–24. [Google Scholar] [CrossRef]
- Van Hove, J.L.K.; Coughlin, C., II; Swanson, M.; Hennermann, J.B. Nonketotic Hyperglycinemia. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef]
- Sacharow, S.J.; Picker, J.D.; Levy, H.L. Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Sahota, A.; Tischfield, J.A.; Goldfarb, D.S.; Ward, M.D.; Hu, L. Cystinuria: Genetic aspects, mouse models, and a new approach to therapy. Urolithiasis 2019, 47, 57–66. [Google Scholar] [CrossRef]
- da Rosa, M.S.; da Rosa-Junior, N.T.; Parmeggiani, B.; Glanzel, N.M.; de Moura Alvorcem, L.; Ribeiro, R.T.; Grings, M.; Wajner, M.; Leipnitz, G. 3-Hydroxy-3-Methylglutaric Acid Impairs Redox and Energy Homeostasis, Mitochondrial Dynamics, and Endoplasmic Reticulum-Mitochondria Crosstalk in Rat Brain. Neurotox. Res. 2020, 37, 314–325. [Google Scholar] [CrossRef]
- da Rosa, M.S.; Scaini, G.; Damiani, A.P.; Longaretti, L.M.; Pereira, M.; Seminotti, B.; Zapelini, H.G.; Schuck, P.F.; Streck, E.L.; de Andrade, V.M.; et al. Evidence that 3-hydroxy-3-methylglutaric and 3-methylglutaric acids induce DNA damage in rat striatum. Metab. Brain Dis. 2015, 30, 1055–1062. [Google Scholar] [CrossRef]
- Duran, M.; Schutgens, R.B.; Ketel, A.; Heymans, H.; Bertssen, M.W.; Ketting, D.; Wadman, S.K. 3-hydroxy-3-methylglutaryl coenzyme A lyase deficiency: Postnatal management following prenatal diagnosis by analysis of maternal urine. J. Pediatric 1979, 95, 1004–1007. [Google Scholar] [CrossRef]
- Gibson, K.M.; Breuer, J.; Kaiser, K.; Nyhan, W.L.; McCoy, E.E.; Ferreira, P.; Greene, C.L.; Blitzer, M.G.; Shapira, E.; Reverte, F.; et al. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Report of five new patients. J. Inherit. Metab. Dis. 1988, 11, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Nadler, J.V.; Wang, A.; Hakim, A. Toxicity of L-proline toward rat hippocampal neurons. Brain Res. 1988, 456, 168–172. [Google Scholar] [CrossRef]
- Ferreira, A.G.; da Cunha, A.A.; Scherer, E.B.; Machado, F.R.; da Cunha, M.J.; Braga, A.; Mussulini, B.H.; Moreira, J.D.; Wofchuk, S.; Souza, D.O.; et al. Evidence that hyperprolinemia alters glutamatergic homeostasis in rat brain: Neuroprotector effect of guanosine. Neurochem. Res. 2012, 37, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Oyanagi, K.; Tsuchiyama, A.; Itakura, Y.; Tamura, Y.; Nakao, T.; Fujita, S.; Shiono, H. Clinical, biochemical and enzymatic studies in type I hyperprolinemia associated with chromosomal abnormality. Tohoku J. Exp. Med. 1987, 151, 465–475. [Google Scholar] [CrossRef]
- Woody, N.C. Hyperlysinemia. Am. J. Dis. Child 1964, 108, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Sharma, G.S.; Singh, L.R. Homocystinuria: Therapeutic approach. Clin. Chim. Acta 2016, 458, 55–62. [Google Scholar] [CrossRef]
- Watkins, D.; Ru, M.; Hwang, H.Y.; Kim, C.D.; Murray, A.; Philip, N.S.; Kim, W.; Legakis, H.; Wai, T.; Hilton, J.F.; et al. Hyperhomocysteinemia due to methionine synthase deficiency, cblG: Structure of the MTR gene, genotype diversity, and recognition of a common mutation, P1173L. Am. J. Hum. Genet. 2002, 71, 143–153. [Google Scholar] [CrossRef]
- Zavadakova, P.; Fowler, B.; Zeman, J.; Suormala, T.; Pristoupilova, K.; Kozich, V.; Zavad’akova, P. CblE type of homocystinuria due to methionine synthase reductase deficiency: Clinical and molecular studies and prenatal diagnosis in two families. J. Inherit. Metab. Dis. 2002, 25, 461–476. [Google Scholar] [CrossRef]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar] [PubMed]
- McCully, K.S. Chemical pathology of homocysteine. I. Atherogenesis. Ann. Clin. Lab. Sci. 1993, 23, 477–493. [Google Scholar] [PubMed]
- Ueland, P.M.; Refsum, H. Plasma homocysteine, a risk factor for vascular disease: Plasma levels in health, disease, and drug therapy. J. Lab. Clin. Med. 1989, 114, 473–501. [Google Scholar] [PubMed]
- Mattson, M.P.; Shea, T.B. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef]
- Sun, C.F.; Haven, T.R.; Wu, T.L.; Tsao, K.C.; Wu, J.T. Serum total homocysteine increases with the rapid proliferation rate of tumor cells and decline upon cell death: A potential new tumor marker. Clin. Chim. Acta 2002, 321, 55–62. [Google Scholar] [CrossRef]
- Zhang, D.; Lou, J.; Zhang, X.; Zhang, L.; Wang, F.; Xu, D.; Niu, N.; Wang, Y.; Wu, Y.; Cui, W. Hyperhomocysteinemia results from and promotes hepatocellular carcinoma via CYP450 metabolism by CYP2J2 DNA methylation. Oncotarget 2017, 8, 15377–15392. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Lombardi, C.; Acanfora, F.; Satta, E.; Cesare, C.M.; Violetti, E.; Romano, M.M.; De Santo, N.G. Possible mechanisms of homocysteine toxicity. Kidney Int. Suppl. 2003, 84, S137–S140. [Google Scholar] [CrossRef]
- Ostrakhovitch, E.A.; Tabibzadeh, S. Homocysteine and age-associated disorders. Ageing Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef]
- Sade Yazdi, D.; Laor Bar-Yosef, D.; Adsi, H.; Kreiser, T.; Sigal, S.; Bera, S.; Zaguri, D.; Shaham-Niv, S.; Oluwatoba, D.S.; Levy, D.; et al. Homocysteine fibrillar assemblies display cross-talk with Alzheimer’s disease beta-amyloid polypeptide. Proc. Natl. Acad. Sci. USA 2021, 118, e2017575118. [Google Scholar] [CrossRef]
- Haavik, J.; Toska, K. Tyrosine hydroxylase and Parkinson’s disease. Mol. Neurobiol. 1998, 16, 285–309. [Google Scholar] [CrossRef] [PubMed]
- van Kessel, S.P.; Frye, A.K.; El-Gendy, A.O.; Castejon, M.; Keshavarzian, A.; van Dijk, G.; El Aidy, S. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat. Commun. 2019, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Koshti, B.; Kshtriya, V.; Singh, R.; Walia, S.; Bhatia, D.; Joshi, K.B.; Gour, N. Unusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine. ACS Chem. Neurosci. 2021, 12, 3237–3249. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, N.; Kim, D. Toxic Metabolites and Inborn Errors of Amino Acid Metabolism: What One Informs about the Other. Metabolites 2022, 12, 527. https://doi.org/10.3390/metabo12060527
Lee N, Kim D. Toxic Metabolites and Inborn Errors of Amino Acid Metabolism: What One Informs about the Other. Metabolites. 2022; 12(6):527. https://doi.org/10.3390/metabo12060527
Chicago/Turabian StyleLee, Namgyu, and Dohoon Kim. 2022. "Toxic Metabolites and Inborn Errors of Amino Acid Metabolism: What One Informs about the Other" Metabolites 12, no. 6: 527. https://doi.org/10.3390/metabo12060527
APA StyleLee, N., & Kim, D. (2022). Toxic Metabolites and Inborn Errors of Amino Acid Metabolism: What One Informs about the Other. Metabolites, 12(6), 527. https://doi.org/10.3390/metabo12060527