
Rifampicin-Mediated Metabolic Changes in Mycobacterium tuberculosis

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
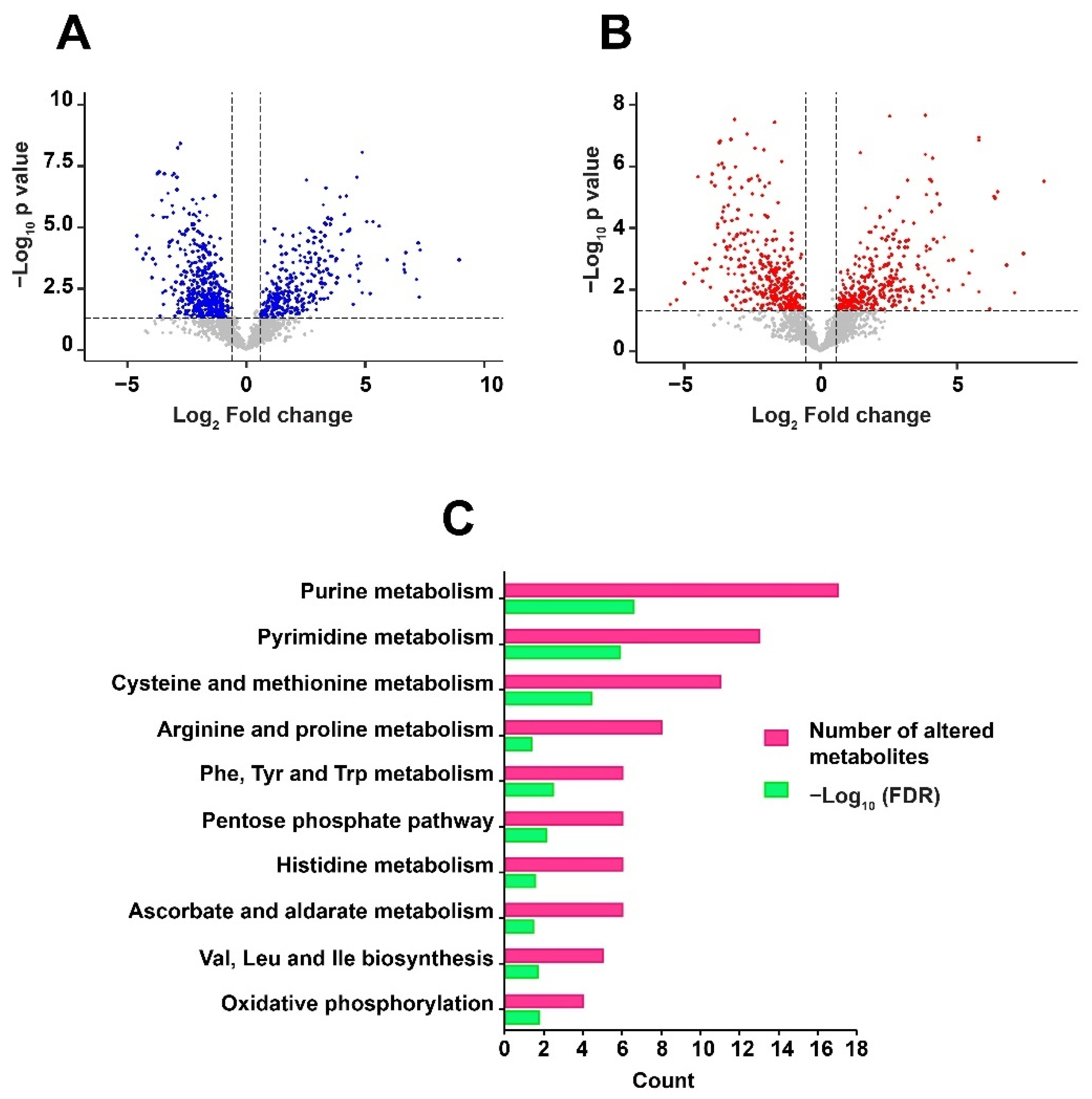
2.1. MS2 Identification of Mtb Metabolites by RIF Treatment
2.2. Metabolic Dysregulation in Mtb by RIF
2.3. Metabolic Pathway Analysis and Classification of Metabolites
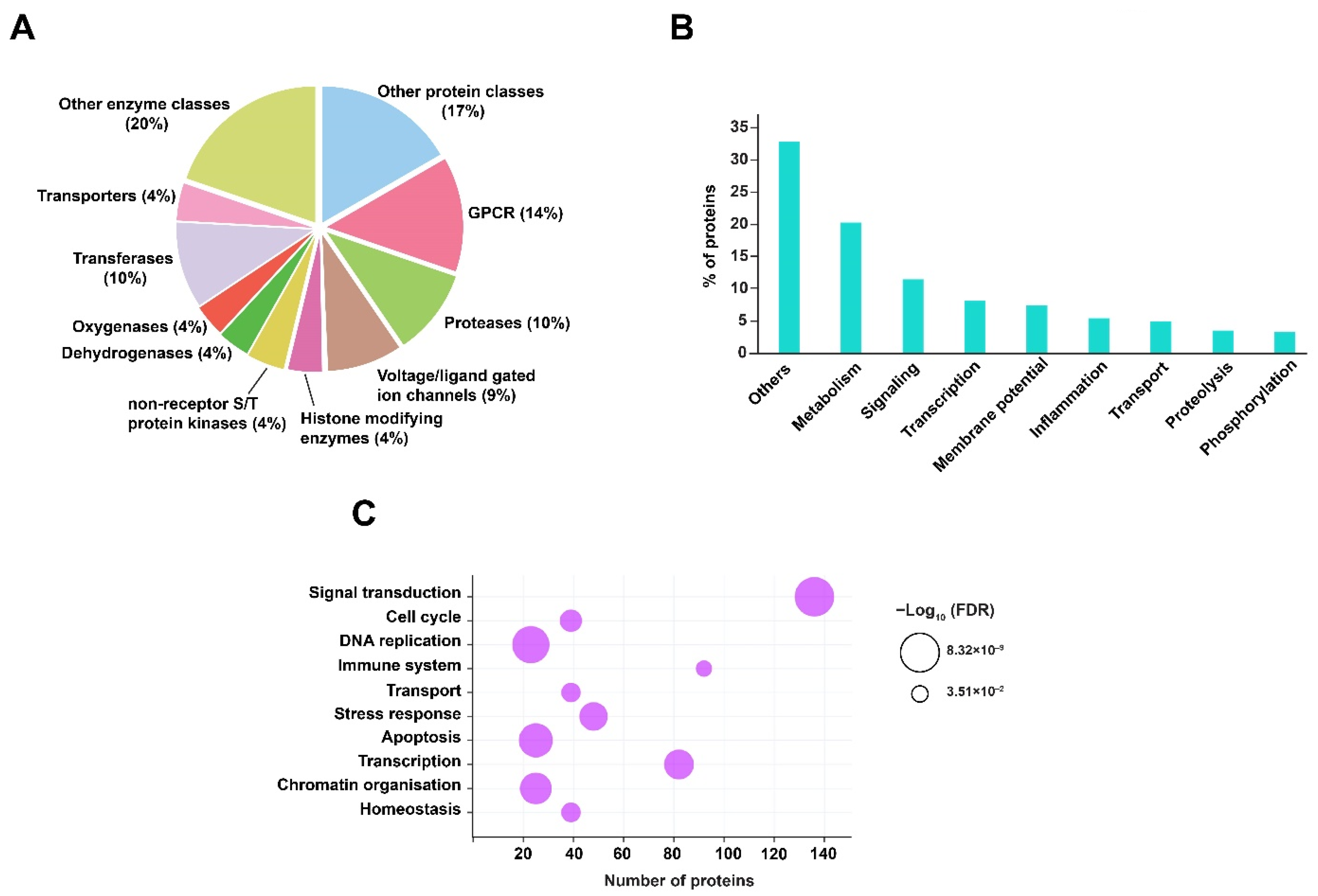
2.4. Prediction of Host Protein Targets against RIF Mediated Mtb Dysregulated Metabolites
2.5. Multiple Reaction Monitoring (MRM)-Based Validation of Altered Metabolites
3. Discussion
4. Materials and Methods
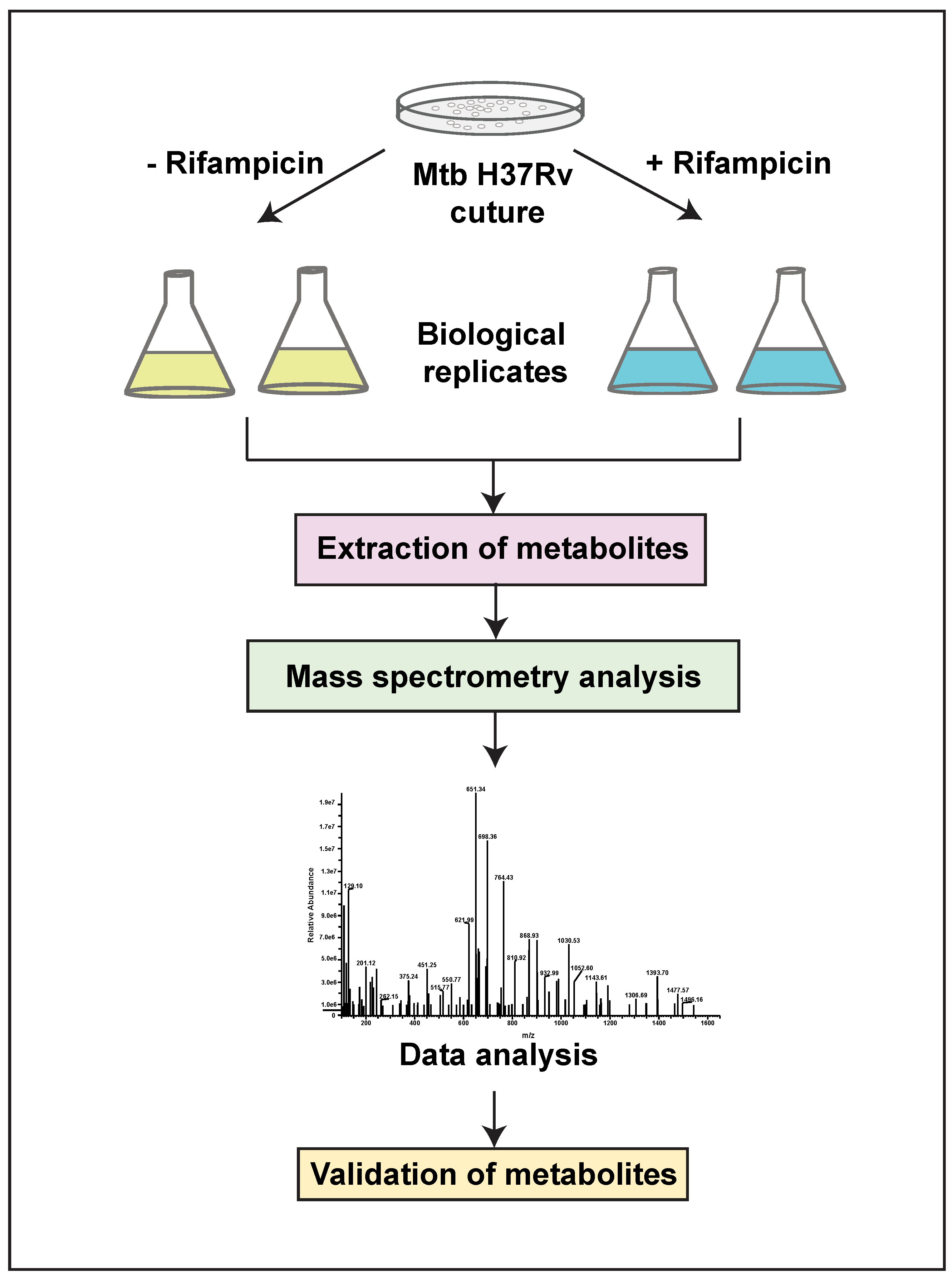
4.1. Bacterial Culture
4.2. Bacterial Cell Lysis and Metabolite Separation
4.3. Tandem Mass Spectrometry Analysis for Untargeted Metabolomics
4.4. Data Analysis and Metabolite Mapping
4.5. Bioinformatic Analysis
4.6. MRM-Based Validation
4.7. Microplate Alamar Blue Assay (MABA)
4.8. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TB | Tuberculosis |
| RIF | Rifampicin |
| PCA | Principal Component Analysis |
| PLS-DA | PLS discriminant analysis |
| FDR | False Discovery Rate |
| GO | Gene Ontology |
| ACN | Acetonitrile |
| MRM | Multiple reaction monitoring |
| TCA | Tricarboxylic acid |
| FC | Fold change |
| QC | Quality control |
| DP | Declustering Potential |
| RT | Retention time |
| CE | Collision Energy |
| EP | Entry Potential |
| CXP | Cell exit potential |
| MABA | Microplate Alamar blue assay |
References
- Yelamanchi, S.D.; Surolia, A. Targeting amino acid metabolism of Mycobacterium tuberculosis for developing inhibitors to curtail its survival. IUBMB Life 2021, 73, 643–658. [Google Scholar] [CrossRef]
- World Health Organisation (WHO). Global Tuberculosis Report 2021; WHO: Geneva, Switzerland, 2021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 7 March 2022).
- Xu, G.; Liu, H.; Jia, X.; Wang, X.; Xu, P. Mechanisms and detection methods of Mycobacterium tuberculosis rifampicin resistance: The phenomenon of drug resistance is complex. Tuberculosis 2021, 128, 102083. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- McClure, W.R.; Cech, C.L. On the mechanism of rifampicin inhibition of RNA synthesis. J. Biol. Chem. 1978, 253, 8949–8956. [Google Scholar] [CrossRef]
- Kessler, C.; Hartmann, G.R. The two effects of rifampicin on the RNA polymerase reaction. Biochem. Biophys. Res. Commun. 1977, 74, 50–56. [Google Scholar] [CrossRef]
- Zaw, M.T.; Emran, N.A.; Lin, Z. Mutations inside rifampicin-resistance determining region of rpoB gene associated with rifampicin-resistance in Mycobacterium tuberculosis. J. Infect. Public Health 2018, 11, 605–610. [Google Scholar] [CrossRef]
- Louw, G.E.; Warren, R.M.; Gey van Pittius, N.C.; Leon, R.; Jimenez, A.; Hernandez-Pando, R.; McEvoy, C.R.; Grobbelaar, M.; Murray, M.; van Helden, P.D.; et al. Rifampicin reduces susceptibility to ofloxacin in rifampicin-resistant Mycobacterium tuberculosis through efflux. Am. J. Respir. Crit. Care Med. 2011, 184, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, M.; Nathan, C.; Rhee, K.Y. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun. 2014, 5, 4306. [Google Scholar] [CrossRef]
- Zhu, J.H.; Wang, B.W.; Pan, M.; Zeng, Y.N.; Rego, H.; Javid, B. Rifampicin can induce antibiotic tolerance in mycobacteria via paradoxical changes in rpoB transcription. Nat. Commun. 2018, 9, 4218. [Google Scholar] [CrossRef] [Green Version]
- Brucoli, F.; McHugh, T.D. Rifamycins: Do not throw the baby out with the bathwater. Is rifampicin still an effective anti-tuberculosis drug? Future Med. Chem. 2021, 13, 2129–2131. [Google Scholar] [CrossRef]
- Sterling, T.R.; Njie, G.; Zenner, D.; Cohn, D.L.; Reves, R.; Ahmed, A.; Menzies, D.; Horsburgh, C.R., Jr.; Crane, C.M.; Burgos, M.; et al. Guidelines for the Treatment of Latent Tuberculosis Infection: Recommendations from the National Tuberculosis Controllers Association and CDC, 2020. MMWR Recomm. Rep. 2020, 69, 1–11. [Google Scholar] [CrossRef]
- Loots, D.T. New insights into the survival mechanisms of rifampicin-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2016, 71, 655–660. [Google Scholar] [CrossRef]
- du Preez, I.; Loots, D.T. Altered fatty acid metabolism due to rifampicin-resistance conferring mutations in the rpoB Gene of Mycobacterium tuberculosis: Mapping the potential of pharmaco-metabolomics for global health and personalized medicine. OMICS 2012, 16, 596–603. [Google Scholar] [CrossRef]
- Bisson, G.P.; Mehaffy, C.; Broeckling, C.; Prenni, J.; Rifat, D.; Lun, D.S.; Burgos, M.; Weissman, D.; Karakousis, P.C.; Dobos, K. Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J. Bacteriol. 2012, 194, 6441–6452. [Google Scholar] [CrossRef] [Green Version]
- Man, D.K.; Kanno, T.; Manzo, G.; Robertson, B.D.; Lam, J.K.W.; Mason, A.J. Rifampin- or Capreomycin-Induced Remodeling of the Mycobacterium smegmatis Mycolic Acid Layer Is Mitigated in Synergistic Combinations with Cationic Antimicrobial Peptides. mSphere 2018, 3, e00218-18. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Marchev, A.S.; Vasileva, L.V.; Amirova, K.M.; Savova, M.S.; Balcheva-Sivenova, Z.P.; Georgiev, M.I. Metabolomics and health: From nutritional crops and plant-based pharmaceuticals to profiling of human biofluids. Cell. Mol. Life Sci. 2021, 78, 6487–6503. [Google Scholar] [CrossRef]
- Zhang, X.W.; Li, Q.H.; Xu, Z.D.; Dou, J.J. Mass spectrometry-based metabolomics in health and medical science: A systematic review. RSC Adv. 2020, 10, 3092–3104. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Paley, S.; Karp, P.D. The BioCyc Metabolic Network Explorer. BMC Bioinform. 2021, 22, 208. [Google Scholar] [CrossRef]
- Martinez-Sena, T.; Luongo, G.; Sanjuan-Herraez, D.; Castell, J.V.; Vento, M.; Quintas, G.; Kuligowski, J. Monitoring of system conditioning after blank injections in untargeted UPLC-MS metabolomic analysis. Sci. Rep. 2019, 9, 9822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashokan, M.; Ramesha, K.P.; Hallur, S.; Karthikkeyan, G.; Rana, E.; Azharuddin, N.; Raj, S.R.; Jeyakumar, S.; Kumaresan, A.; Kataktalware, M.A.; et al. Differences in milk metabolites in Malnad Gidda (Bos indicus) cows reared under pasture-based feeding system. Sci. Rep. 2021, 11, 2831. [Google Scholar] [CrossRef] [PubMed]
- Saraf, K.K.; Kumaresan, A.; Dasgupta, M.; Karthikkeyan, G.; Prasad, T.S.K.; Modi, P.K.; Ramesha, K.; Jeyakumar, S.; Manimaran, A. Metabolomic fingerprinting of bull spermatozoa for identification of fertility signature metabolites. Mol. Reprod. Dev. 2020, 87, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ibanez, J.; Pazos, F.; Chagoyen, M. MBROLE 2.0-functional enrichment of chemical compounds. Nucleic Acids Res. 2016, 44, W201–W204. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Piccaro, G.; Pietraforte, D.; Giannoni, F.; Mustazzolu, A.; Fattorini, L. Rifampin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 7527–7533. [Google Scholar] [CrossRef] [Green Version]
- Cumming, B.M.; Chinta, K.C.; Reddy, V.P.; Steyn, A.J.C. Role of Ergothioneine in Microbial Physiology and Pathogenesis. Antioxid. Redox Signal. 2018, 28, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Ung, K.S.; Av-Gay, Y. Mycothiol-dependent mycobacterial response to oxidative stress. FEBS Lett. 2006, 580, 2712–2716. [Google Scholar] [CrossRef] [Green Version]
- Van Laer, K.; Buts, L.; Foloppe, N.; Vertommen, D.; Van Belle, K.; Wahni, K.; Roos, G.; Nilsson, L.; Mateos, L.M.; Rawat, M.; et al. Mycoredoxin-1 is one of the missing links in the oxidative stress defence mechanism of Mycobacteria. Mol. Microbiol. 2012, 86, 787–804. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Brzostek, A.; Pawelczyk, J.; Rumijowska-Galewicz, A.; Dziadek, B.; Dziadek, J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 2009, 191, 6584–6591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casabon, I.; Crowe, A.M.; Liu, J.; Eltis, L.D. FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol. Microbiol. 2013, 87, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Fontes, F.L.; Gonzalez-Juarrero, M.; McNeil, M.R.; Crans, D.C.; Jackson, M.; Crick, D.C. Partial Saturation of Menaquinone in Mycobacterium tuberculosis: Function and Essentiality of a Novel Reductase, MenJ. ACS Cent. Sci. 2015, 1, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.K.; Mahapatra, S.; Slayden, R.A.; Boyne, M.E.; Lenaerts, A.; Hinshaw, J.C.; Angala, S.K.; Chatterjee, D.; Biswas, K.; Narayanasamy, P.; et al. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol. 2009, 72, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukheja, P.; Kumar, P.; Mittal, N.; Li, S.G.; Singleton, E.; Russo, R.; Perryman, A.L.; Shrestha, R.; Awasthi, D.; Husain, S.; et al. A Novel Small-Molecule Inhibitor of the Mycobacterium tuberculosis Demethylmenaquinone Methyltransferase MenG Is Bactericidal to Both Growing and Nutritionally Deprived Persister Cells. mBio 2017, 8, e02022-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meganathan, R.; Kwon, O. Biosynthesis of Menaquinone (Vitamin K2) and Ubiquinone (Coenzyme Q). EcoSal Plus 2009, 3. [Google Scholar] [CrossRef] [Green Version]
- Dosselaere, F.; Vanderleyden, J. A metabolic node in action: Chorismate-utilizing enzymes in microorganisms. Crit. Rev. Microbiol. 2001, 27, 75–131. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
- Gil, D.; Garcia, L.F.; Rojas, M. Modulation of macrophage apoptosis by antimycobacterial therapy: Physiological role of apoptosis in the control of Mycobacterium tuberculosis. Toxicol. Appl. Pharmacol. 2003, 190, 111–119. [Google Scholar] [CrossRef]
- Sharma, R.; Muttil, P.; Yadav, A.B.; Rath, S.K.; Bajpai, V.K.; Mani, U.; Misra, A. Uptake of inhalable microparticles affects defence responses of macrophages infected with Mycobacterium tuberculosis H37Ra. J. Antimicrob. Chemother. 2007, 59, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Smirnov, A.; Pluskal, T.; Jia, W.; Sumner, S. Metabolomics Data Preprocessing Using ADAP and MZmine 2. Methods Mol. Biol. 2020, 2104, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Yelamanchi, S.D.; Arun Kumar, S.T.; Mishra, A.; Keshava Prasad, T.S.; Surolia, A. Metabolite Dysregulation by Pranlukast in Mycobacterium tuberculosis. Molecules 2022, 27, 1520. [Google Scholar] [CrossRef] [PubMed]
- Behera, S.K.; Kasaragod, S.; Karthikkeyan, G.; Narayana Kotimoole, C.; Raju, R.; Prasad, T.S.K.; Subbannayya, Y. MS2Compound: A User-Friendly Compound Identification Tool for LC-MS/MS-Based Metabolomics Data. OMICS 2021, 25, 389–399. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Pon, A.; Karu, N.; Zheng, J.; Li, C.; Arndt, D.; Gautam, M.; Allen, F.; Wishart, D.S. CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 2019, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.E.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Adams, K.J.; Pratt, B.; Bose, N.; Dubois, L.G.; St John-Williams, L.; Perrott, K.M.; Ky, K.; Kapahi, P.; Sharma, V.; MacCoss, M.J.; et al. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19, 1447–1458. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | Metabolite | Mode of Acquisition | Fold Change | p-Value |
|---|---|---|---|---|
| 1 | 2C-Methyl-D-erythritol 2,4-cyclodiphosphate | Positive | 3.33 | 0.01 |
| 2 | L-Glutamine | Positive | 48.68 | 0.00 |
| 3 | Thymidine | Negative | 0.47 | 0.02 |
| 4 | Thymidine-5′-phosphate | Negative | 0.10 | 0.00 |
| 5 | Uridine-5′-diphosphate | Positive | 5.42 | 0.01 |
| 6 | Deoxycytidine diphosphate | Positive | 0.09 | 0.00 |
| 7 | 2-Isopropylmaleic acid | Positive | 1.70 | 0.03 |
| 8 | 3-Deoxy-D-arabino-heptulosonate-7-phosphate | Positive | 14.13 | 0.00 |
| 9 | 4-Guanidinobutyric acid | Positive | 5.93 | 0.02 |
| 10 | Biotin | Negative | 2.14 | 0.02 |
| 11 | Cyclic AMP | Negative | 9.25 | 0.00 |
| 12 | Histidinal | Positive | 1.83 | 0.00 |
| 13 | L-Cystathionine | Negative | 3.95 | 0.03 |
| 14 | Menaquinone-9 | Negative | 0.19 | 0.00 |
| 15 | S-Adenosyl-L-homocysteine | Negative | 4.09 | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yelamanchi, S.D.; Mishra, A.; Behra, S.K.; Karthikkeyan, G.; Keshava Prasad, T.S.; Surolia, A. Rifampicin-Mediated Metabolic Changes in Mycobacterium tuberculosis. Metabolites 2022, 12, 493. https://doi.org/10.3390/metabo12060493
Yelamanchi SD, Mishra A, Behra SK, Karthikkeyan G, Keshava Prasad TS, Surolia A. Rifampicin-Mediated Metabolic Changes in Mycobacterium tuberculosis. Metabolites. 2022; 12(6):493. https://doi.org/10.3390/metabo12060493
Chicago/Turabian StyleYelamanchi, Soujanya D., Archita Mishra, Santosh Kumar Behra, Gayathree Karthikkeyan, Thottethodi Subrahmanya Keshava Prasad, and Avadhesha Surolia. 2022. "Rifampicin-Mediated Metabolic Changes in Mycobacterium tuberculosis" Metabolites 12, no. 6: 493. https://doi.org/10.3390/metabo12060493
APA StyleYelamanchi, S. D., Mishra, A., Behra, S. K., Karthikkeyan, G., Keshava Prasad, T. S., & Surolia, A. (2022). Rifampicin-Mediated Metabolic Changes in Mycobacterium tuberculosis. Metabolites, 12(6), 493. https://doi.org/10.3390/metabo12060493