Abstract
Early research has suggested a rather straightforward relation between phosphate exposure, increased serum FGF23 (Fibroblast Growth Factor 23) concentrations and clinical endpoints. Unsurprisingly, however, subsequent studies have revealed a much more complex interplay between autocrine and paracrine factors locally in bone like PHEX and DMP1, concentrations of minerals in particular calcium and phosphate, calciprotein particles, and endocrine systems like parathyroid hormone PTH and the vitamin D system. In addition to these physiological regulators, an expanding list of disease states are shown to influence FGF23 levels, usually increasing it, and as such increase the burden of disease. While some of these physiological or pathological factors, like inflammatory cytokines, may partially confound the association of FGF23 and clinical endpoints, others are in the same causal path, are targetable and hence hold the promise of future treatment options to alleviate FGF23-driven toxicity, for instance in chronic kidney disease, the FGF23-associated disease with the highest prevalence by far. These factors will be reviewed here and their relative importance described, thereby possibly opening potential means for future therapeutic strategies.
Keywords:
FGF23 (Fibroblast Growth Factor 23); regulation; mineral metabolism; PTH; DMP1; phosphate; calcium; vitamin D 1. Introduction
Fibroblast Growth Factor 23 (FGF23) has emerged as an important biomarker in chronic kidney disease (CKD) [1]. Accumulating evidence suggests that it not only is a risk predictor for cardiovascular disease, in particular heart disease and heart failure, but also a uremic toxin itself, directly causing disease [2]. For both properties, being either an independent risk predictor or a direct toxin, in-depth knowledge of its regulation is of paramount importance. In the setting of FGF23 as an independent risk factor, but not directly inflicting harm, its association with clinical endpoints is confounded by hitherto hidden mechanisms that are in the causal path to these endpoints. Exploring these regulators of FGF23 may thus reveal novel targets of treatment and hold the promise of improving outcomes for patients with kidney disease. In turn, if FGF23 itself is the causative molecule, intervening in its regulators may also modify FGF23-driven morbidity.
Besides being a prominent hormone in CKD, the discovery of FGF23 solved the quest for a humoral factor explaining several inheritable diseases characterized by renal wasting, which by then could be explained by mutations of FGF23 itself or factors involved in its regulation [3].
FGF23 is a hormone, secreted by osteocytes, and has a central physiological role in phosphate homeostasis. It promotes phosphaturia and inhibits the activation of vitamin D, thereby limiting vitamin-D mediated phosphate absorption from the diet by the transcellular uptake route of enterocytes in the gastro-intestinal tract. There are several principal ways in which FGF23 concentrations can be regulated, and all of these appear to play a role. These modes of regulation are production and secretion by the cells of origin, ectopic production, and cleavage or breakdown at cells of origin or after release into the circulation. The currently available immunoassays measure either the full-length and biologically active hormone, termed intact FGF23 (iFGF23), or both iFGF23 and its c-terminal fragment, termed cFGF23. It should be noted that the term cFGF23 for this assay is a confusing term, because it does not only measure the c-terminal fragment, the latter originating after cleavage of the full-length polypeptide. This cleavage obviously also generates an N-terminal fragment, but no commercially available assay detects this fraction. Although intact FGF23 is assumed to be the physiological effector molecule, debate exists on the role of the fragments, possibly having agonistic or antagonistic effects, the latter as a competitive inhibitor [4].
2. The Role of Minerals as Regulators of FGF23
2.1. Phosphate
Given the key role of FGF23 to protect the organism against hyperphosphatemia, it can be expected that phosphate increases FGF23 concentrations. Indeed, several studies have shown that an increase in dietary intake of phosphate by both healthy volunteers and people with CKD increased its concentrations, albeit with some delay of around 24 h [5,6,7] to restore phosphate balance. In turn, phosphate restriction has the ability to lower FGF23, but, different from PTH secretion from healthy parathyroid glands in a setting of hypercalcemia, FGF23 has never been described to be fully suppressed following hypophosphatemia, for instance when induced by mutations in the renal phosphate transporter NaPi2c which gives rise to hereditary hypophosphatemic rickets with hypercalciuria (HHRH) [8,9]. Until recently, the underlying molecular mechanism by which phosphate modulates FGF23 levels has been elusive. It has been shown that phosphate transport into bone cells across the inorganic phosphate transporter 1 (PiT-1) may be involved [10]. A recent study revealed an additional remarkable mechanism [11]. Bone cells that produce FGF23 express its receptor FGFR1 as well. It is now shown that phosphate itself can bind to this unliganded receptor, leading to the upregulation of the Galnt3 gene, the protein-product of which leads to O-glycosylation of full-length FGF23, as will be discussed below. The consequence of this post-translational modification of the FGF23 molecule is that it escapes intracellular cleavage, increasing the proportion of biologically active FGF23. This mechanism does not suggest that phosphate induces FGF23 expression, even though a previous study suggested it can in a cell line [12], but rather stabilises the hormone. This mode of action of phosphate on FGF23 concentrations is in line with clinical studies in patients with CKD that addressed the question of whether dietary phosphate restriction can lower FGF23. A recent meta-analysis of these studies found more pronounced reduction of iFGF23 than of cFGF23, the latter also measuring FGF23 fragments [13]. In normophosphatemic CKD patients, short-term treatment with non-calcium containing phosphate binders did not change FGF23 [14,15], while prolonged treatment induced a substantial decline [16]. The use of calcium-containing binders did increase FGF23 [17].
2.2. Calcium
Interestingly, there appears to be a minimal concentration of calcium required for phosphate to be able to increase FGF23 levels. In an animal model testing varying serum concentration of calcium, it was shown that an increment of FGF23 by PTH was completely abolished when ionized calcium concentrations were below 4 mg/dL [18]. The physiological functionality of this phenomenon might be that this prevents the catabolism of vitamin D by FGF23 in a setting of hypocalcemia. Moreover, in an animal model, calcium itself was shown to be able to directly increase FGF23 transcription by acting on the promotor of the Fgf23 gene [19,20]. These findings from experimental research are in line with most, but not all, clinical observations. In a clinical trial among 30 early CKD patients, studying the effects of adding calcium carbonate to calcitriol, it was shown that this induced an increase of FGF23, which was paralleled by an increase in serum calcium concentration [21]. In more advanced CKD, the non-calcium containing phosphate binder lanthanum carbonate was able to lower FGF23 levels, while a calcium-containing binder could not [17]. However, in a short-term study, acute increments or decrements of serum calcium concentrations had no effect on FGF23 [22].
2.3. Calciprotein Particles
Apart from the synergistic effects of combined higher levels of calcium and phosphate on increasing FGF23, it is possible that this is mediated by the formation of calciprotein particles (CPP) [23,24]. Even at physiological concentrations, human plasma is supersaturated for calcium and phosphate, which would induce spontaneous hydroxyapatite crystal formation [25]. These potentially damaging crystals, however, are prevented from being formed and freely floating in the circulation by being scavenged into soluble amorphous primary calciprotein particles CPP (CPP1), which are nanoparticles containing the serum protein Fetuin-A as the main protein constituent. In a setting of increased availability of these minerals, as is the case for phosphate in CKD, or suppressed hepatic production of Fetuin A in a setting of chronic inflammation, the stage is set to overwhelm the capacity of this defence system, leading to the formation of more toxic larger crystalline secondary CPPs (CPP2) [26,27,28]. Like high exposure to phosphate, exposure to high calcium levels also increases the amount of CPP, as was shown in a patient with renal sarcoidosis, and this was paralleled by an increase in FGF23 [29]. The role of calcium on the formation of CPP was also shown in a clinical study comparing calciumcarbonate with lanthanumcarbonate [30]. After switching to lanthanumcarbonate, the total amount of CPP declined substantially, without major differences in serum concentration of calcium and phosphate between the two phosphate binders.
A recent clinical observational study demonstrated an association between the amount of CPPs and FGF23, suggesting an induction in the phosphaturic hormone by CPP’s [31]. Indeed, a recent in vitro study found that CPPs are capable of increasing FGF23 expression in osteoblast-like cells [32]. Remarkably, this effect was restricted to the smaller sized CPP1. It is therefore conceivable that an increased amount of CPP’s formed triggers FGF23, which in turn induces phosphaturia and declines levels of active vitamin D. FGF23 thereby slows the formation of CPP’s by lowering the concentrations of the minerals that form its mineral content. This concept is supported by the ability of CPP to exit the circulation, enter the bone marrow and reach FGF23-producing bone cells [32].
2.4. Magnesium
Given its resemblance to calcium as a bivalent cation, and its beneficial effects on the formation of CPP [33,34,35], it is likely that magnesium is also involved in the regulation of FGF23. Data, however, are scarce. In an animal study of cats with chronic kidney disease, a negative association between serum magnesium concentration and FGF23 was found, which was independent of calcium, phosphate, and PTH [36]. In an observational study among young healthy men, it was shown that a lower dietary intake of magnesium was associated with higher FGF23 [37]. When rats were exposed to a short-term (7 day) magnesium deficient diet, FGF23 levels were higher compared to a normal diet at all time points following the interventions which reached statistical significance after one week [38]. However, clinical trials demonstrating a beneficial effect on clinically relevant endpoints of magnesium supplements are lacking [39].
The role of minerals and calciprotein particles are summarized in Figure 1.
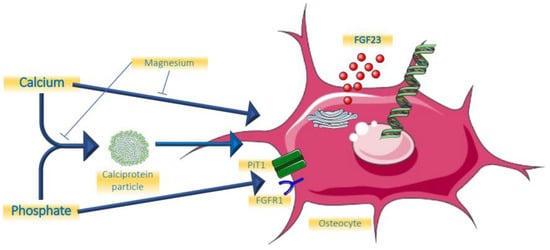
Figure 1.
Effects of minerals on FGF23.
3. Hormonal Regulation of FGF23
3.1. Parathyroid Hormone
PTH was shown to be a relevant regulator of FGF23 by directly increasing its expression in bone in an experimental model of CKD [40]. Moreover, in that same study, parathyroidectomy before the onset of CKD completely abolished the FGF23 increment, even in a subsequent setting of hyperphosphatemia. This is probably mediated by the receptor PTH1R for PTH on bone cells, the same receptor that is involved in regulating bone turnover, and with Nuclear Receptor Related-1 protein (Nurr1) an intermediate intracellular molecule [41]. Another established action of PTH on bone cells is the suppression of the gene encoding for sclerostin (Sost). Sclerostin acts as local inhibitor of the Wnt pathway by sclerostin, thereby suppressing FGF23 [42,43,44]. PTH therefore unleashes FGF23 by suppressing sclerostin. Clinical studies suggest a biphasic response to PTH. In a short term (3 h) 1–34 PTH infusion in healthy young persons, FGF23 declined, most likely driven by PTH-induced renal phosphate loss [45]. During this period 1,25 dihydroxyvitamin D3 (1,25D) started to rise, which expectedly would induce increased dietary phosphate uptake. This, and the potential direct effects of PTH on bone cells may be the dominating effect following more prolonged exposure, giving rise to FGF23 increments. This indeed was suggested by a two days PTH infusion study that led to increased cFGF23 in healthy persons and people treated by dialysis regardless of bone turnover status [46]. Like for many other aspects, however, the role of PTH is complex, because if endogenous levels rise as a consequence of a decline of serum calcium by sodium citrate infusion, FGF23 did not increase [22]. Obviously, the stimulating effects of PTH on FGF23 may have been nullified by the low levels of calcium. There seems to be a logical physiological basis for the induction of FGF23 by PTH. The key purpose of PTH is to restore hypocalcemia and it does so in part by liberating calcium form bone. This is paralleled by release of phosphate, which is, besides by phosphaturic effects of PTH itself, excreted by the kidneys under the influence of FGF23.
Observations of persons with dialysis-dependent end-stage kidney disease treated with calcimimetics appear to be in line with the notion that lowering PTH is accompanied by declining FGF23 [47,48]. Remarkably, however, in both of these clinical studies, using the oral cinacalcet or the intravenous etelcalcetide, the decline of FGF23 followed reductions of phosphate and calcium, instead of PTH reductions.
3.2. Vitamin D
There is strong evidence that 1,25D directly induces Fgf23 gene transcription. Mice injected with the active form of vitamin D had increased levels of FGF23 mRNA, exclusively in bone, which was accompanied by a rise in serum FGF23 levels [49]. In that same study, rat-derived UMR-106 osteoblast-like cells had a 1000-fold increase of FGF23 mRNA 4 h after exposure to 1,25D. In another study with a focus on exploring the Fgf23 gene promotor region, this role of 1,25D was confirmed [50]. Collins and co-workers observed three patients that received a high dose of calcitriol after parathyroidectomy after surgery and observed steep increments of FGF23 [51]. Many clinical trials have been performed in which either active or nutritional vitamin D was the key intervention. In several of these trials, FGF23 levels were part of the follow-up parameters. The results of these observations have been summarized in two meta-analyses. In the first of these it was found that in patients that were deficient in vitamin D at baseline, the intervention induced a statistically significant increase of iFGF23 [52]. There was also an increase of cFGF23, but this did not reach statistical significance. A very recent meta-analysis could not confirm this effect of vitamin D, but in this meta-analysis, trials were included where participants did not have vitamin D deficiency at baseline, which may explain the discrepancy with the previous analysis [53]. In a study among children treated by dialysis, active vitamin D compounds (calcitriol or doxercalciferol) induced a substantial increase in FGF23 [54]. Collectively, these studies strongly suggest that vitamin D, especially active vitamin D, induces FGF23.
A summary of the roles of PTH and vitamin D is provided in Figure 2.
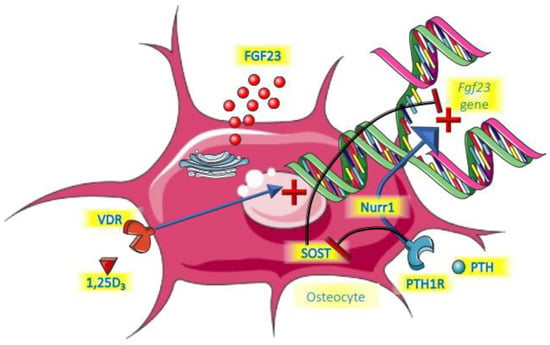
Figure 2.
Endocrine control of FGF23.
4. Local Regulators of FGF23 in Bone
4.1. Factors Involved in FGF23 Expression
Dentin Matrix Protein 1 (DMP1) and PHosphate regulating gene with homologies to Endopeptidases on the X chromosome (PHEX) both are suppressors of FGF23 gene expression that appear to act in concert locally in bone for that function [55,56,57]. PHEX is also believed to promote FGF23 cleavage, which then would induce a lower iFGF23 over cFGF23 ratio. Mutations in either PHEX (XLH, X-linked hypophosphatemic rickets) and DMP1 (ARHR, autosomal recessive hypophosphatemic rickets) cause renal phosphate wasting and its clinical sequelae by primary elevations of FGF23. There are no descriptions in the literature of acquired malfunction or suppression of the PHEX protein, with the possible exception of a report on a patient with leprosy [58]. For DMP1, however, diseases that induce acquired suppression appear to exist. In a mice model of CKD, it was shown that renal failure lowered osteocyte DMP1 expression, followed by FGF23 increases, while supplementation of DMP1 partially restored FGF23 towards the normal lower range [59]. The extent to which this is of relevance in clinical CKD remains to be established, but it has been shown that lower circulating levels of DMP1 are associated with cardiovascular event [60], and this finding may be mediated by increases of FGF23. In addition, uremia induced suppression of DMP1, and hence increments of FGF23 may explain the clinical observation that in more early stages of CKD, FGF23 and phosphate levels appear to diverge, pointing to another inducer of FGF23 than phosphate itself, namely suppressed DMP1 [61]. α-Klotho is intricately involved in phosphate homeostasis and the biological activity of FGF23 [1]. Its colocalization with FGFR1 is mandatory for signal transduction of FGF23 across the cell membrane to exert its actions in the proximal tubules, to induce phosphaturia. Recent research has now revealed that the circulating form of α-klotho, generated after cleavage of its large ectodomain [62], is involved in the expression and excretion of FGF23 from osteocytes. This hitherto unknown role of α-klotho was postulated after analysis of a 13-months old girl with unexplained elevation of FGF23 leading to hypophosphatemic rickets [63]. She was found to have a translocation nearby the α-klotho gene. This phenotype could be mimicked in an animal model by using an adenovector-induced increased expression of α-klotho, leading to high levels of circulating α-klotho, accompanied by a very steep rise of FGF23 and hypophosphatemia [64]. A recent study employed targeted deletion of the α-klotho gene from long bones and found that this led to attenuated increase of FGF23 after induction of CKD, both at osteocyte expression level and its circulating concentration [65]. This strongly suggests that α-klotho is required in an autocrine fashion for FGF23 expression from osteocytes. Both studies revealed that the presence of FGFR1 on osteocytes is required.
4.2. Post-Translational Modification of FGF23 in Bone
Following the translation of FGF23, the full-length polypeptide can be cleaved intracellularly before being secreted, thereby preventing the biologically active compound to enter the circulation. This cleavage occurs between the arginine residues at positions 176 and 179, and mutations at either of the arginine residues renders FGF23 resistant to proteolytic cleavage, giving rise to autosomal dominant hypophosphatemic rickets (ADHR) [66]. This cleavage is assumed to occur at the Golgi-apparatus by one of seven serine-proteases belonging to the family of subtilisin-like preprotein convertases (SPC), which act by cleaving polypeptides from preproteins to its mature polypeptide backbone. The most likely SPC is furin because its knock-out completely prevented FGF23 cleavage [67]. Prior to being exposed to these proteases, in particular furin, FGF23 can be O-glycosylated by N-acetylgalactosaminyltransferase 3 (GalNT3) at threonine residue position 178, which induces resistance to proteolytic cleavage of FGF23. As indicated above, exposure to phosphate may increase this O-glycosylation and thereby increase the relative amount of full-length FGF23, the active form, as a feedback mechanism to restore phosphate to lower concentrations. In turn, FGF23 can also be phosphorylated at a serine residue at position180 by a kinase termed Fam20c, which prohibits O-glycosylation by GalNT3, which ultimately makes FGF23 more prone for proteolytic cleavage [67].
5. Clinical Conditions and Their Impact of FGF23
5.1. Anemia and Iron Deficiency
Patients with ADHR, one of the inherited forms of renal phosphate wasting due to inappropriate elevations of FGF23, can present rather late (from puberty or not even before their mid-forties), and frequently do not present with typical features such as short stature or bowed deformations of the lower extremity [68]. While these patients have limited or absent capacity to cleave FGF23, it is assumed that as long as the baseline transcription of FGF23 is rather low, circulating iFGF23 can remain relatively normal for years without severe phosphate losses. Iron deficiency in these patients was associated with increased iFGF23 levels [69] and in a small open label trial oral iron supplementation substantially lowered FGF23 level in patients with ADHR [70]. These clinical observations are in line with animal research on models of ADHR [71]. In that experimental study it was additionally shown that exposure of osteoblastic cells (UMR-106) to low iron condition increased mRNA of FGF23 up to 20-fold. The mechanisms involved were mitogen-activated protein kinase (MAPK) dependent. In addition, iron-deficiency also induced increments of Hypoxia Inducible Factor 1α (HIF1α), and HIF1α itself could also boost FGF23 expression. Indeed, it was shown that a HIF1α binding site exists in the promotor region of the Fgf23 gene [72]. In addition HIF1α prevents the cleavage of FGF23 [73]. Collectively, these findings would lead to higher circulation levels of iFGF23 in a setting of increased expression of HIF1α by either iron deficiency or hypoxia. However, in a study using the HIF1α stabiliser molidustat in an animal model of CKD and in additional in vitro experiments, it was shown that improved iron availability to osteocytes by the compound abolished the increased FGF23 expression [74]. This same study also revealed that EPO increased FGF23 [74], and this finding was previously shown in both patients and animal models [75]. This latter study demonstrated that this effect was sustained after bone marrow ablation, where upregulation of the Fgf23 gene persisted, strongly suggesting a direct effect on these cells in cortical bone. Also in human studies, either EPO levels or exogenous doses were associated with FGF23, in particular total FGF23, while the effects on iFGF23 were indeterminant [76]. The role of hepcidin, a liver-derived acute phase protein that induces functional iron deficiency, as an intermediate metabolite in anemia, and iron-deficiency associated FGF23 upregulation is not yet well established.
5.2. Inflammation
Several reports point to the role of inflammatory mediators on bone cells leading to increased expression and secretion of FGF23 [73,77]. In turn, FGF23 can upregulate inflammatory mediators from hepatocytes [77]. Especially in the setting of advanced CKD with remarkably high concentrations of FGF23, this may initiate a pro-inflammatory vicious circle, further driving FGF23. Indeed, several pro-inflammatory cytokines such as tumor necrosis factor (TNF), Interleukin-1β (IL-1β), TNF-like weak inducers of apoptosis (TWEAK) and also bacterial lipopolysaccahrides (LPS) have been shown to stimulate both Fgf23 gene expression and protein excretion in a cell model of osteocytes [78]. In another study, LPS injections increased FGF23 despite a low phosphate diet [79]. Interestingly, the exposure to LPS also caused renal FGF23 resistance by suppression of kidney α-klotho, thereby dismantling the FGF23 receptor.
Using several animal models of CKD, or TNF injections in mice with normal kidney function, it was found that TNF increased FGF23 while anti-TNF prevented this [80]. Importantly, the source of FGF23 in that study was the kidney itself, possibly driven by the highest local concentrations of TNF in that organ. This role of TNF is in line with the identification of a TNF responsive FGF23 enhancer, suggesting the direct upregulation of FGF23 by this inflammatory cytokine [81], although it has also been suggested that increases of NF-κB are required.
5.3. Chronic Kidney Disease
An extensive review of the impact of chronic kidney disease on FGF23 is beyond the scope of this review and has been extensively reviewed recently [1,82]. Besides the propensity to accumulate phosphate as a driver for FGF23 increases, in addition to hyperparathyroidism, DMP1 suppression, as outlined above, chronic inflammation, iron deficiency, and FGF23 resistance due to α-klotho deficiency have all been implicated in the exponential rise of FGF23 as CKD progresses. Importantly, experimental studies found that FGF23 cleavage in CKD is impaired as it is in ADHR [73,83]. This feature of CKD, the precise molecular mechanisms of which is currently unknown, fits with the observation that in end stage kidney disease, most circulating FGF23 is intact [84]. Interestingly, it was recently shown that in a model of acute kidney injury, the kidneys themselves produce glycerol-3-phosphate (G3P), which directly stimulates FGF23 production, exclusively in bone [85]. It is likely that besides novel regulators like G3P, the impact of CKD on many, if not all, of the mechanisms involved, as described above, is huge, and collectively creates a perfect storm for essentially unopposed upregulation of FGF23. In addition, it seems plausible that in the setting of CKD, the cleavage of FGF23 is attenuated or its capacity overwhelmed, leading to extremely high levels of biologically active FGF23 in end stage kidney disease, most likely contributing to uremic toxicity.
6. Conclusions
The physiological regulation of bone-derived FGF23 is complex, and is regulated at levels of gene transcription, post-translational modifications, cleavage and cellular release. In addition, remote biological activity is variable by dynamic affinity of its receptor due to changing α-klotho abundance, possibly competitive inhibition by FGF23-fragments, and also varying expression of the FGF receptors themselves [86]. Moreover, ectopic FGF23 production has been described too, as outlined for the kidney as described above, but cardiac production has also been described [87,88]. The machinery involved in regulating the metabolism of FGF23 involves an intricate interplay between minerals, calciprotein particles, the endocrine system and local regulators in the vicinity of osteoblasts and osteocytes in an autocrine or paracrine fashion. Since FGF23 is most likely involved in the pathogenesis of an expanding list of diseases, in-depth knowledge of these regulatory pathways is the first step in ultimately targeting these molecular mechanisms that are in the path to clinical events. The exploration of these pathways is far from being finalized, and designing safe and effective interventions are only at the beginning.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Vervloet, M. Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 2018, 15, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Bouma-de Krijger, A.; Vervloet, M.G. Fibroblast growth factor 23: Are we ready to use it in clinical practice? J. Nephrol. 2020, 33, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Aljuraibah, F.; Bacchetta, J.; Brandi, M.L.; Florenzano, P.; Javaid, M.K.; Mäkitie, O.; Raimann, A.; Rodriguez, M.; Siggelkow, H.; Tiosano, D.; et al. An Expert Perspective on Phosphate Dysregulation With a Focus on Chronic Hypophosphatemia. J. Bone Miner. Res. 2022, 37, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef]
- Vervloet, M.G.; van Ittersum, F.J.; Buttler, R.M.; Heijboer, A.C.; Blankenstein, M.A.; ter Wee, P.M. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin. J. Am. Soc. Nephrol. 2011, 6, 383–389. [Google Scholar] [CrossRef]
- Sigrist, M.; Tang, M.; Beaulieu, M.; Espino-Hernandez, G.; Er, L.; Djurdjev, O.; Levin, A. Responsiveness of FGF-23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): Results of a randomized trial. Nephrol. Dial. Transplant. 2013, 28, 161–169. [Google Scholar] [CrossRef]
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524. [Google Scholar] [CrossRef]
- Tavana, N.; Thilakavathy, K.; Kennerson, M.L.; Ting, T.H. Genetic basis of hereditary hypophosphataemic rickets and phenotype presentation in children and adults. Endokrynol. Pol. 2021, 72, 366–394. [Google Scholar] [CrossRef]
- Gattineni, J.; Baum, M. Genetic disorders of phosphate regulation. Pediatr. Nephrol. 2012, 27, 1477–1487. [Google Scholar] [CrossRef]
- Bon, N.; Frangi, G.; Sourice, S.; Guicheux, J.; Beck-Cormier, S.; Beck, L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol. Metab. 2018, 11, 197–204. [Google Scholar] [CrossRef]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2016, 34, 132–139. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Wu, H.-Y.; Peng, Y.-S.; Hsu, S.-P.; Chiu, Y.-L.; Chen, H.-Y.; Yang, J.-Y.; Ko, M.-J.; Pai, M.-F.; Tu, Y.-K.; et al. Effects of lower versus higher phosphate diets on fibroblast growth factor-23 levels in patients with chronic kidney disease: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2018, 33, 1977–1983. [Google Scholar] [CrossRef]
- Bouma-de Krijger, A.; van Ittersum, F.J.; Hoekstra, T.; Ter Wee, P.M.; Vervloet, M.G. Short-term effects of sevelamer-carbonate on fibroblast growth factor 23 and pulse wave velocity in patients with normophosphataemic chronic kidney disease Stage 3. Clin. Kidney J. 2019, 12, 678–685. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Lu, J.L.; Wall, B.M.; Gyamlani, G.; Naseer, A.; Wallick, A.; Han, Z.; Thomas, F.; Quarles, L.D.; Jarmukli, N. Changes With Lanthanum Carbonate, Calcium Acetate, and Phosphorus Restriction in CKD: A Randomized Controlled Trial. Kidney Int. Rep. 2018, 3, 897–904. [Google Scholar] [CrossRef]
- Ketteler, M.; Sprague, S.M.; Covic, A.C.; Rastogi, A.; Spinowitz, B.; Rakov, V.; Walpen, S.; Floege, J. Effects of sucroferric oxyhydroxide and sevelamer carbonate on chronic kidney disease-mineral bone disorder parameters in dialysis patients. Nephrol. Dial. Transplant. 2018, 34, 1163–1170. [Google Scholar] [CrossRef]
- Soriano, S.; Ojeda, R.; Rodríguez, M.; Almadén, Y.; Rodriguez, M.; Martín-Malo, A.; Aljama, P. The effect of phosphate binders, calcium and lanthanum carbonate on FGF23 levels in chronic kidney disease patients. Clin. Nephrol. 2013, 80, 17–22. [Google Scholar] [CrossRef]
- Rodriguez-Ortiz, M.E.; Lopez, I.; Muñoz-Castañeda, J.R.; Martínez, J.; Peralta-Ramírez, A.; Pineda, C.; Canalejo, A.; Jaeger, P.; Aguilera-Tejero, E.; Rodriguez, M.; et al. Calcium deficiency reduces circulating levels of FGF23. J. Am. Soc. Nephrol. 2012, 23, 1190–1197. [Google Scholar] [CrossRef]
- David, V.; Dai, B.; Martin, A.; Huang, J.; Han, X.; Quarles, L.D. Calcium regulates FGF-23 expression in bone. Endocrinology 2013, 154, 4469–4482. [Google Scholar] [CrossRef]
- Shikida, Y.; Mizobuchi, M.; Inoue, T.; Hamada, T.; Ogata, H.; Koiwa, F.; Shibata, T. Effect of Continuous Intravenous Calcium Loading on Fibroblast Growth Factor 23 in Normal and Uremic Rats. Calcif. Tissue Int. 2018, 103, 455–464. [Google Scholar] [CrossRef]
- Han, N.; Hong, S.H.; Kim, Y.S.; Kim, D.K.; Kim, I.-W.; Ji, E.; Oh, J.M. Effect of additive calcium administration on FGF23 levels in patients with mild chronic kidney disease treated with calcitriol: A randomized, open-labeled clinical trial. Ther. Clin. Risk Manag. 2017, 13, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Wesseling-Perry, K.; Wang, H.; Elashoff, R.; Gales, B.; Jüppner, H.; Salusky, I.B. Lack of FGF23 response to acute changes in serum calcium and PTH in humans. J. Clin. Endocrinol. Metab. 2014, 99, E1951–E1956. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Pipich, V.; Jahnen-Dechent, W.; Schwahn, D. Fetuin-A is a mineral carrier protein: Small angle neutron scattering provides new insight on Fetuin-A controlled calcification inhibition. Biophys. J. 2010, 99, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.G.; Smith, E.R. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol. Dial. Transplant. 2016, 31, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Gangneux, C.; Daveau, M.; Hiron, M.; Derambure, C.; Papaconstantinou, J.; Salier, J.P. The inflammation-induced down-regulation of plasma Fetuin-A (alpha2HS-Glycoprotein) in liver results from the loss of interaction between long C/EBP isoforms at two neighbouring binding sites. Nucleic Acids Res. 2003, 31, 5957–5970. [Google Scholar] [CrossRef][Green Version]
- Mutluay, R.; Değertekin, C.K.; Sayilar, E.I.; Derici, Ü.; Gültekin, S.; Gönen, S.; Arinsoy, S.T. Serum fetuin-A is associated with the components of MIAC(malnutrition, inflammation, atherosclerosis, calcification) syndrome in different stages of chronic kidney disease. Turk. J. Med. Sci. 2019, 49, 327–335. [Google Scholar] [CrossRef]
- Cozzolino, M.; Galassi, A.; Biondi, M.L.; Turri, O.; Papagni, S.; Mongelli, N.; Civita, L.; Gallieni, M.; Brancaccio, D. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am. J. Nephrol. 2006, 26, 423–429. [Google Scholar] [CrossRef]
- Iwazu, Y.; Kuro, O.M.; Miura, Y.; Takeda, S.I.; Yamada, T.; Nagata, D. Calciprotein particles and fibroblast growth factor 23 contribute to the pathophysiology of hypercalcemia in a patient with renal sarcoidosis. Clin. Kidney J. 2021, 14, 421–423. [Google Scholar] [CrossRef]
- Nakamura, K.; Nagata, Y.; Hiroyoshi, T.; Isoyama, N.; Fujikawa, K.; Miura, Y.; Matsuyama, H.; Kuro-O, M. The effect of lanthanum carbonate on calciprotein particles in hemodialysis patients. Clin. Exp. Nephrol. 2020, 24, 323–329. [Google Scholar] [CrossRef]
- Miura, Y.; Iwazu, Y.; Shiizaki, K.; Akimoto, T.; Kotani, K.; Kurabayashi, M.; Kurosu, H.; Kuro-O, M. Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Sci. Rep. 2018, 8, 1256. [Google Scholar] [CrossRef]
- Akiyama, K.-I.; Miura, Y.; Hayashi, H.; Sakata, A.; Matsumura, Y.; Kojima, M.; Tsuchiya, K.; Nitta, K.; Shiizaki, K.; Kurosu, H.; et al. Calciprotein particles regulate fibroblast growth factor-23 expression in osteoblasts. Kidney Int. 2020, 97, 702–712. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Schou, M.; Pasch, A.; Brandi, L. The Effect of Increasing Dialysate Magnesium on Serum Calcification Propensity in Subjects with End Stage Kidney Disease: A Randomized, Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2018, 13, 1373–1380. [Google Scholar] [CrossRef]
- Pasch, A.; Farese, S.; Gräber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-based test measures overall propensity for calcification in serum. J. Am. Soc. Nephrol. 2012, 23, 1744–1752. [Google Scholar] [CrossRef]
- Moor, M.B.; Ramakrishnan, S.K.; Legrand, F.; Bachtler, M.; Koesters, R.; Hynes, N.E.; Pasch, A.; Bonny, O. Elevated serum magnesium lowers calcification propensity in Memo1-deficient mice. PLoS ONE 2020, 15, e0236361. [Google Scholar] [CrossRef]
- van den Broek, D.H.N.; Chang, Y.M.; Elliott, J.; Jepson, R.E. Prognostic importance of plasma total magnesium in a cohort of cats with azotemic chronic kidney disease. J. Vet. Intern. Med. 2018, 32, 1359–1371. [Google Scholar] [CrossRef]
- Kosk, D.; Kramer, H.; Luke, A.; Camacho, P.; Bovet, P.; Rhule, J.P.; Forrester, T.; Wolf, M.; Sempos, C.; Melamed, M.L.; et al. Dietary factors and fibroblast growth factor-23 levels in young adults with African ancestry. J. Bone Miner. Metab. 2017, 35, 666–674. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Katsumata, S.; Maeda, Y.; Kajita, Y. Changes in circulating levels of fibroblast growth factor 23 induced by short-term dietary magnesium deficiency in rats. Magnes. Res. 2016, 29, 48–54. [Google Scholar] [CrossRef]
- Leenders, N.H.J.; Vervloet, M.G. Magnesium: A Magic Bullet for Cardiovascular Disease in Chronic Kidney Disease? Nutrients 2019, 11, 455. [Google Scholar] [CrossRef]
- Lavi-Moshayoff, V.; Wasserman, G.; Meir, T.; Silver, J.; Naveh-Many, T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am. J. Physiol. Ren. Physiol. 2010, 299, F882–F889. [Google Scholar] [CrossRef]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Liang, Y.; Göransson, O.; Sundberg, T.B.; Wang, J.; Williams, E.A.; O’Meara, M.J.; Govea, N.; Beqo, B.; Nishimori, S.; et al. SIKs control osteocyte responses to parathyroid hormone. Nat. Commun. 2016, 7, 13176. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Imanishi, Y.; Tateishi, T.; Miyaoka, D.; Kurajoh, M.; Arnold, A.; Emoto, M. Parathyroid Hormone Regulates Circulating Levels of Sclerostin and FGF23 in a Primary Hyperparathyroidism Model. J. Endocr. Soc. 2022, 6, bvac027. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.A.; Davison, R.; Shakthivel, S.; Anderson, K.D.; Ko, F.C.; Ross, R.D. Sclerostin antibody improves phosphate metabolism hormones, bone formation rates, and bone mass in adult Hyp mice. Bone 2022, 154, 116201. [Google Scholar] [CrossRef]
- Gutierrez, O.M.; Smith, K.T.; Barchi-Chung, A.; Patel, N.M.; Isakova, T.; Wolf, M. (1-34) Parathyroid hormone infusion acutely lowers fibroblast growth factor 23 concentrations in adult volunteers. Clin. J. Am. Soc. Nephrol. 2012, 7, 139–145. [Google Scholar] [CrossRef][Green Version]
- Wesseling-Perry, K.; Harkins, G.C.; Wang, H.-J.; Elashoff, R.; Gales, B.; Horwitz, M.J.; Stewart, A.F.; Jüppner, H.; Salusky, I.B. The calcemic response to continuous parathyroid hormone (PTH)(1-34) infusion in end-stage kidney disease varies according to bone turnover: A potential role for PTH(7-84). J. Clin. Endocrinol. Metab. 2010, 95, 2772–2780. [Google Scholar] [CrossRef]
- Koizumi, M.; Komaba, H.; Nakanishi, S.; Fujimori, A.; Fukagawa, M. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol. Dial. Transplant. 2012, 27, 784–790. [Google Scholar] [CrossRef]
- Wolf, M.; Block, G.A.; Chertow, G.M.; Cooper, K.; Fouqueray, B.; Moe, S.M.; Sun, Y.; Tomlin, H.; Vervloet, M.; Oberbauer, R. Effects of etelcalcetide on fibroblast growth factor 23 in patients with secondary hyperparathyroidism receiving hemodialysis. Clin. Kidney J. 2020, 13, 75–84. [Google Scholar] [CrossRef]
- Kolek, O.I.; Hines, E.R.; Jones, M.D.; LeSueur, L.K.; Lipko, M.A.; Kiela, P.R.; Collins, J.F.; Haussler, M.R.; Ghishan, F.K. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: The final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1036–G1042. [Google Scholar] [CrossRef]
- Ito, M.; Sakai, Y.; Furumoto, M.; Segawa, H.; Haito, S.; Yamanaka, S.; Nakamura, R.; Kuwahata, M.; Miyamoto, K.-I. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1101–E1109. [Google Scholar] [CrossRef]
- Collins, M.T.; Lindsay, J.R.; Jain, A.; Kelly, M.H.; Cutler, C.M.; Weinstein, L.S.; Liu, J.; Fedarko, N.; Winer, K.K. Fibroblast growth factor-23 is regulated by 1alpha,25-dihydroxyvitamin D. J. Bone Miner. Res. 2005, 20, 1944–1950. [Google Scholar] [CrossRef]
- Charoenngam, N.; Rujirachun, P.; Holick, M.F.; Ungprasert, P. Oral vitamin D3 supplementation increases serum fibroblast growth factor 23 concentration in vitamin D-deficient patients: A systematic review and meta-analysis. Osteoporos. Int. 2019, 30, 2183–2193. [Google Scholar] [CrossRef]
- Meshkini, F.; Soltani, S.; Clark, C.C.; Tam, V.; Meyre, D.; Toupchian, O.; Saraf-Bank, S.; Abdollahi, S. The effect of vitamin D supplementation on serum levels of fibroblast growth factor- 23: A systematic review and meta-analysis of randomized controlled trials. J. Steroid Biochem. Mol. Biol. 2022, 215, 106012. [Google Scholar] [CrossRef]
- Wesseling-Perry, K.; Pereira, R.C.; Sahney, S.; Gales, B.; Wang, H.-J.; Elashoff, R.; Jüppner, H.; Salusky, I.B. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int. 2011, 79, 112–119. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Li, H.; Dai, B.; Feng, J.Q.; Quarles, L.D. Overexpression of the DMP1 C-terminal fragment stimulates FGF23 and exacerbates the hypophosphatemic rickets phenotype in Hyp mice. Mol. Endocrinol. 2012, 26, 1883–1895. [Google Scholar] [CrossRef]
- Martin, A.; Liu, S.; David, V.; Li, H.; Karydis, A.; Feng, J.Q.; Quarles, L.D. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. 2011, 25, 2551–2562. [Google Scholar] [CrossRef]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Silva, S.R.B.; Illarramendi, X.; Tempone, A.J.; Silva, P.H.L.; Nery, J.A.C.; Monteiro, A.M.V.; Pessolani, M.C.V.; Boasquevisque, E.; Sarno, E.N.; Pereira, G.M.B.; et al. Downregulation of PHEX in multibacillary leprosy patients: Observational cross-sectional study. J. Transl. Med. 2015, 13, 296. [Google Scholar] [CrossRef]
- Dussold, C.; Gerber, C.; White, S.; Wang, X.; Qi, L.; Francis, C.; Capella, M.; Courbon, G.; Wang, J.; Li, C.; et al. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone Res. 2019, 7, 12. [Google Scholar] [CrossRef]
- Martin, A.; Kentrup, D. The Role of DMP1 in CKD-MBD. Curr. Osteoporos. Rep. 2021, 19, 500–509. [Google Scholar] [CrossRef]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Amin, R.; Moe, O.W.; Hu, M.C.; Erben, R.G.; Ostman Wernerson, A. The kidney is the principal organ mediating klotho effects. J. Am. Soc. Nephrol. 2014, 25, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.A.; Adler, F.; Nelson-Williams, C.; Iijima, J.; Li, P.; Imura, A.; Nabeshima, Y.-I.; Reyes-Mugica, M.; Carpenter, T.O.; Lifton, R.P. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc. Natl. Acad. Sci. USA 2008, 105, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.C.; O’Bryan, L.M.; Farrow, E.G.; Summers, L.J.; Clinkenbeard, E.L.; Roberts, J.L.; Cass, T.A.; Saha, J.; Broderick, C.; Ma, Y.L.; et al. Circulating alphaKlotho influences phosphate handling by controlling FGF23 production. J. Clin. Investig. 2012, 122, 4710–4715. [Google Scholar] [CrossRef]
- Kaludjerovic, J.; Komaba, H.; Sato, T.; Erben, R.G.; Baron, R.; Olauson, H. Klotho expression in long bones regulates FGF23 production during renal failure. FASEB J. 2017, 31, 2050–2064. [Google Scholar] [CrossRef]
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pages, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Appaiah, H.N.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef]
- Econs, M.J.; McEnery, P.T. Autosomal dominant hypophosphatemic rickets/osteomalacia: Clinical characterization of a novel renal phosphate-wasting disorder. J. Clin. Endocrinol. Metab. 1997, 82, 674–681. [Google Scholar] [CrossRef]
- Imel, E.A.; Peacock, M.; Gray, A.K.; Padgett, L.R.; Hui, S.L.; Econs, M.J. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J. Clin. Endocrinol. Metab. 2011, 96, 3541–3549. [Google Scholar] [CrossRef]
- Imel, E.A.; Liu, Z.; Coffman, M.; Acton, D.; Mehta, R.; Econs, M.J. Oral Iron Replacement Normalizes Fibroblast Growth Factor 23 in Iron-Deficient Patients With Autosomal Dominant Hypophosphatemic Rickets. J. Bone Miner. Res. 2020, 35, 231–238. [Google Scholar] [CrossRef]
- Farrow, E.G.; Yu, X.; Summers, L.J.; Davis, S.I.; Fleet, J.C.; Allen, M.R.; Robling, A.G.; Stayrook, K.R.; Jideonwo, V.; Magers, M.J.; et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1146–E1155. [Google Scholar] [CrossRef]
- Zhang, Q.; Doucet, M.; Tomlinson, R.; Han, X.; Quarles, L.D.; Collins, M.T.; Clemens, T.L. The hypoxia-inducible factor-1alpha activates ectopic production of fibroblast growth factor 23 in tumor-induced osteomalacia. Bone Res. 2016, 4, 16011. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef]
- Noonan, M.; Clinkenbeard, E.L.; Ni, P.; Swallow, E.A.; Tippen, S.P.; Agoro, R.; Allen, M.R.; White, K.E. Erythropoietin and a hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHDi) lowers FGF23 in a model of chronic kidney disease (CKD). Physiol. Rep. 2020, 8, e14434. [Google Scholar] [CrossRef]
- Clinkenbeard, E.L.; Hanudel, M.R.; Stayrook, K.R.; Appaiah, H.N.; Farrow, E.G.; Cass, T.A.; Summers, L.J.; Ip, C.S.; Hum, J.M.; Thomas, J.C.; et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica 2017, 102, e427–e430. [Google Scholar] [CrossRef]
- Hanudel, M.; Eisenga, M.; Rappaport, M.; Chua, K.J.; Qiao, B.; Jung, C.-L.; Gabayan, V.; Gales, B.; Ramos, G.; Jong, M.; et al. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol. Dial. Transplant. 2019, 34, 2057–2065. [Google Scholar] [CrossRef]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef]
- Ito, N.; Wijenayaka, A.R.; Prideaux, M.; Kogawa, M.; Ormsby, R.T.; Evdokiou, A.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol. Cell. Endocrinol. 2015, 399, 208–218. [Google Scholar] [CrossRef]
- Rodríguez-Ortiz, M.E.; Díaz-Tocados, J.M.; Muñoz-Castañeda, J.R.; Herencia, C.; Pineda, C.; Martínez, J.; De Oca, A.M.; López-Baltanás, R.; Alcala-Diaz, J.F.; Ortiz, A.; et al. Inflammation both increases and causes resistance to FGF23 in normal and uremic rats. Clin. Sci. 2020, 134, 15–32. [Google Scholar] [CrossRef]
- Egli-Spichtig, D.; Imenez Silva, P.H.; Glaudemans, B.; Gehring, N.; Bettoni, C.; Zhang, M.Y.H.; Pastor-Arroyo, E.M.; Schönenberger, D.; Rajski, M.; Hoogewijs, D.; et al. Tumor necrosis factor stimulates fibroblast growth factor 23 levels in chronic kidney disease and non-renal inflammation. Kidney Int. 2019, 96, 890–905. [Google Scholar] [CrossRef]
- Onal, M.; Carlson, A.H.; Thostenson, J.D.; Benkusky, N.A.; Meyer, M.B.; Lee, S.M.; Pike, J.W. A Novel Distal Enhancer Mediates Inflammation-, PTH-, and Early Onset Murine Kidney Disease-Induced Expression of the Mouse Fgf23 Gene. JBMR Plus 2018, 2, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G. FGF23 measurement in chronic kidney disease: What is it really reflecting? Clin. Chim. Acta 2020, 505, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Hanudel, M.R.; Chua, K.; Rappaport, M.; Gabayan, V.; Valore, E.; Goltzman, D.; Ganz, T.; Nemeth, E.; Salusky, I.B. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild-type and hepcidin knockout mice. Am. J. Physiol. Ren. Physiol. 2016, 311, F1369–F1377. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Urakawa, I.; Isakova, T.; Yamazaki, Y.; Epstein, M.; Wesseling-Perry, K.; Wolf, M.; Salusky, I.B.; Juüppner, H. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J. Clin. Endocrinol. Metab. 2010, 95, 578–585. [Google Scholar] [CrossRef]
- Simic, P.; Kim, W.; Zhou, W.; Pierce, K.A.; Chang, W.; Sykes, D.B.; Aziz, N.B.; Elmariah, S.; Ngo, D.; Pajevic, P.D.; et al. Glycerol-3-phosphate is an FGF23 regulator derived from the injured kidney. J. Clin. Investig. 2020, 130, 1513–1526. [Google Scholar] [CrossRef]
- Leifheit-Nestler, M.; Grabner, A.; Hermann, L.; Richter, B.; Schmitz, K.; Fischer, D.C.; Yanucil, C.; Faul, C.; Haffner, D. Vitamin D treatment attenuates cardiac FGF23/FGFR4 signaling and hypertrophy in uremic rats. Nephrol. Dial. Transplant. 2017, 32, 1493–1503. [Google Scholar] [CrossRef]
- Matsui, I.; Oka, T.; Kusunoki, Y.; Mori, D.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Kubota, K.; Yonemoto, S.; et al. Cardiac hypertrophy elevates serum levels of fibroblast growth factor 23. Kidney Int. 2018, 94, 60–71. [Google Scholar] [CrossRef]
- Andrukhova, O.; Slavic, S.; Odorfer, K.I.; Erben, R.G. Experimental Myocardial Infarction Upregulates Circulating Fibroblast Growth Factor-23. J. Bone Miner. Res. 2015, 30, 1831–1839. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).