
Rewiring of the Liver Transcriptome across Multiple Time-Scales Is Associated with the Weight Loss-Independent Resolution of NAFLD Following RYGB

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
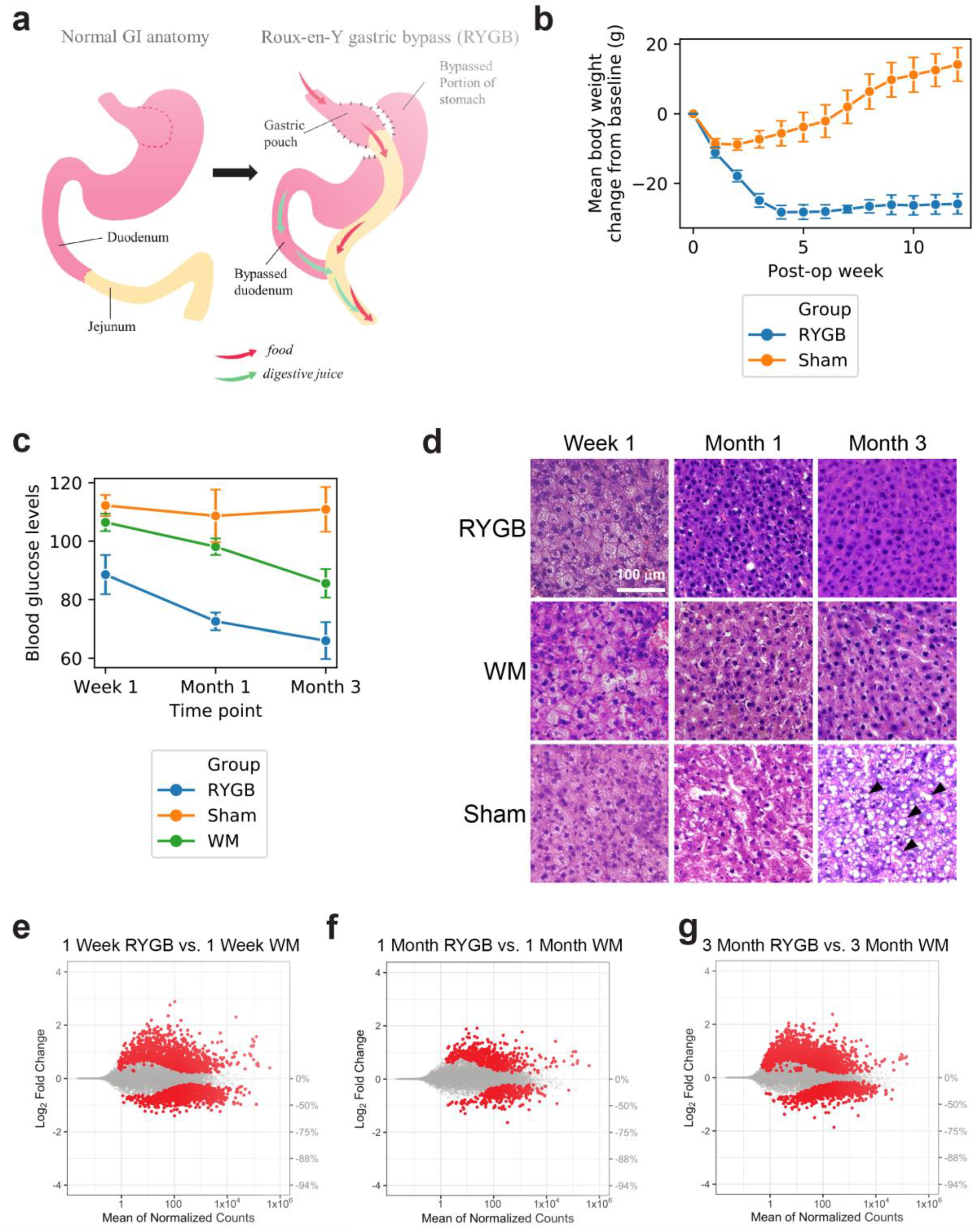
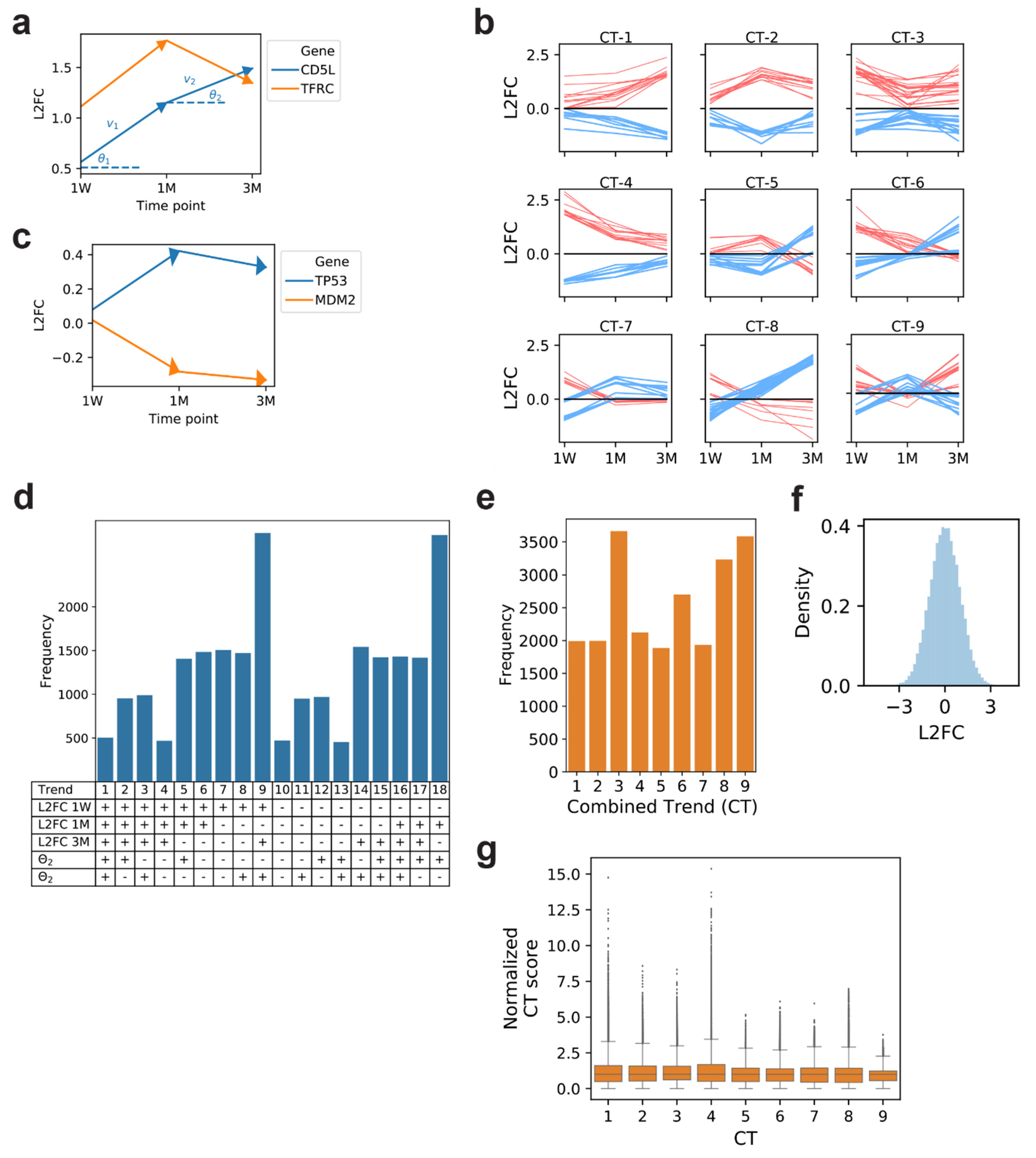
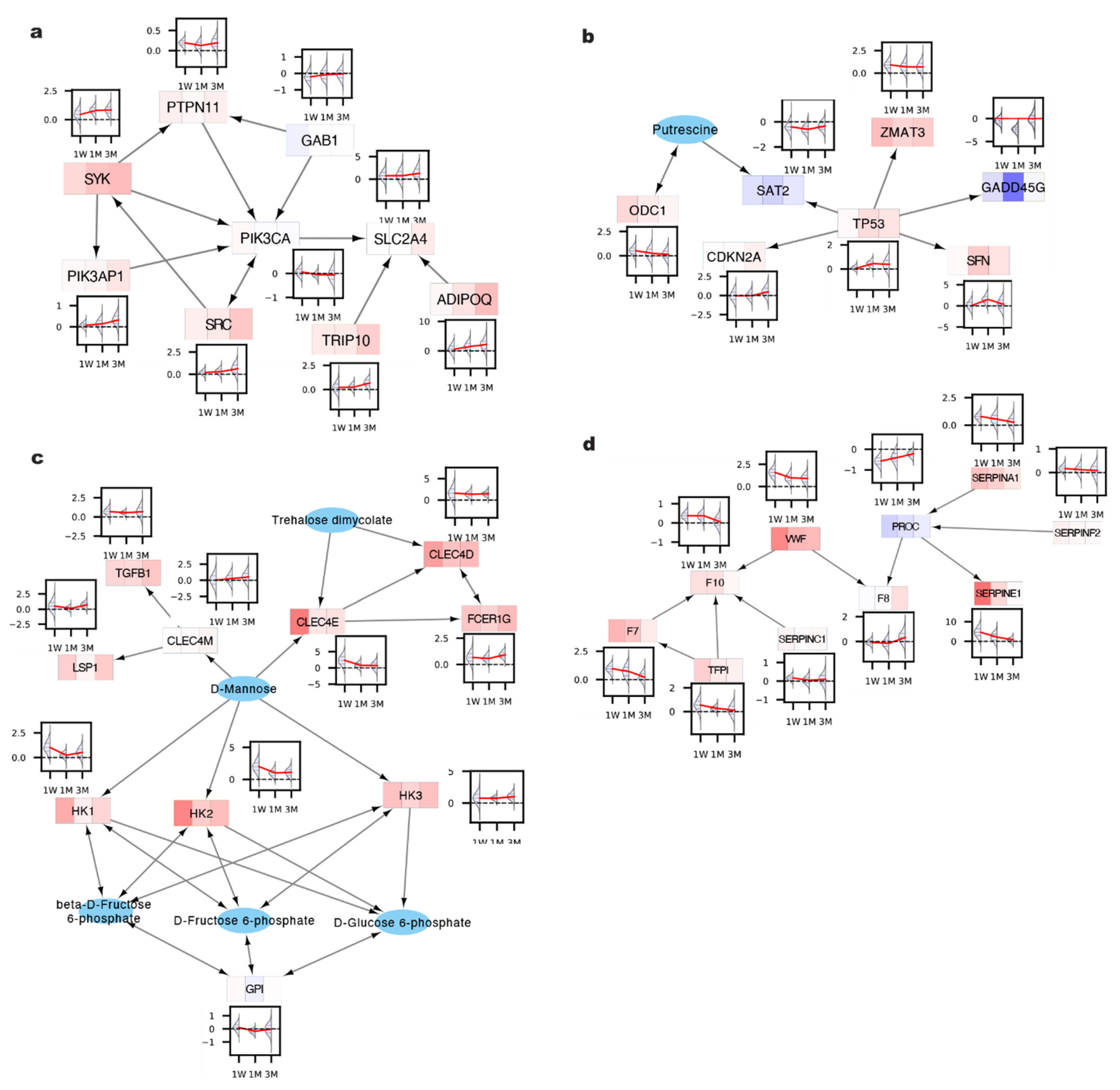
2. Results and Discussion
3. Materials and Methods
3.1. Animals
3.2. Roux-en-Y Gastric Bypass (RYGB) and Sham Surgery
3.3. Intraperitoneal Glucose Tolerance Test
3.4. RNA-seq
3.5. Combined Trends
3.6. Gene CT Scores
3.7. KEGG Human Network (KEGG-HN)
3.8. Generating KEGG-HN Subnetworks
3.9. Module Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ben-Zvi, D.; Meoli, L.; Abidi, W.M.; Nestoridi, E.; Panciotti, C.; Castillo, E.; Pizarro, P.; Shirley, E.; Gourash, W.F.; Thompson, C.C.; et al. Time-Dependent Molecular Responses Differ between Gastric Bypass and Dieting but Are Conserved Across Species. Cell Metab. 2018, 28, 310–323.e6. [Google Scholar] [CrossRef] [Green Version]
- Albrechtsen, N.J.W.; Geyer, P.E.; Doll, S.; Treit, P.V.; Bojsen-Møller, K.N.; Martinussen, C.; Jørgensen, N.B.; Torekov, S.; Meier, F.; Niu, L.; et al. Plasma Proteome Profiling Reveals Dynamics of Inflammatory and Lipid Homeostasis Markers after Roux-En-Y Gastric Bypass Surgery. Cell Syst. 2018, 7, 601–612.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, T.D.; Davidson, L.E.; Litwin, S.E.; Kim, J.; Kolotkin, R.; Nanjee, M.N.; Gutierrez, J.M.; Frogley, S.J.; Ibele, A.R.; Brinton, E.A.; et al. Weight and Metabolic Outcomes 12 Years after Gastric Bypass. N. Engl. J. Med. 2017, 377, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Doumouras, A.G.; Yu, J.; Brar, K.; Banfield, L.; Gmora, S.; Anvari, M.; Hong, D. Complete Resolution of Nonalcoholic Fatty Liver Disease After Bariatric Surgery: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 1040–1060.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michna, A.; Braselmann, H.; Selmansberger, M.; Dietz, A.; Heß, J.; Gomolka, M.; Hornhardt, S.; Blüthgen, N.; Zitzelsberger, H.; Unger, K. Natural Cubic Spline Regression Modeling Followed by Dynamic Network Reconstruction for the Identification of Radiation-Sensitivity Gene Association Networks from Time-Course Transcriptome Data. PLoS ONE 2016, 11, e0160791. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; A Gutiérrez-Pabello, J.; Kramnik, I.; Maiti, T.; Quackenbush, J. An improved empirical bayes approach to estimating differential gene expression in microarray time-course data: BETR (Bayesian Estimation of Temporal Regulation). BMC Bioinform. 2009, 10, 409. [Google Scholar] [CrossRef] [Green Version]
- Äijö, T.; Butty, V.; Chen, Z.; Salo, V.; Tripathi, S.; Burge, C.B.; Lahesmaa, R.; Lähdesmäki, H. Methods for time series analysis of RNA-seq data with application to human Th17 cell differentiation. Bioinformatics 2014, 30, i113–i120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, D.S.; Theis, F.J.; Yosef, N. Impulse model-based differential expression analysis of time course sequencing data. Nucleic Acids Res. 2018, 46, e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straube, J.; Huang, B.E.; Cao, K.-A.L. DynOmics to identify delays and co-expression patterns across time course experiments. Sci. Rep. 2017, 7, 40131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmush, M.L.; D’Alessandro, M.; Saeidi, N. Regulation of Energy Homeostasis after Gastric Bypass Surgery. Annu. Rev. Biomed. Eng. 2017, 19, 459–484. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Adams, R.D.; Saltiel, A.R. The TC10-interacting protein CIP4/2 is required for insulin-stimulated Glut4 translocation in 3T3L1 adipocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 12835–12840. [Google Scholar] [CrossRef] [Green Version]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [Green Version]
- Kaser, S.; Moschen, A.; Cayon, A.; Kaser, A.; Crespo, J.; Pons-Romero, F.; Ebenbichler, C.; Patsch, J.R.; Tilg, H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Krisenko, M.O.; Geahlen, R.L. Calling in SYK: SYK’s dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Biophys. Acta 2015, 1853, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, I.; Ezra, O.; Rivlin, N.; Molchadsky, A.; Madar, S.; Goldfinger, N.; Rotter, V. p53, a novel regulator of lipid metabolism pathways. J. Hepatol. 2012, 56, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Porteiro, B.; Fondevila, M.F.; Buque, X.; Gonzalez-Rellan, M.J.; Fernandez, U.; Mora, A.; Beiroa, D.; Senra, A.; Gallego, R.; Ferno, J.; et al. Pharmacological stimulation of p53 with low-dose doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis. Mol. Metab. 2018, 8, 132–143. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Yang, H.P.; Ward, K.K.; Sahasrabuddhe, V.V.; McGlynn, K.A. Bariatric Surgery and Liver Cancer in a Consortium of Academic Medical Centers. Obes. Surg. 2016, 26, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopec, A.K.; Abrahams, S.R.; Thornton, S.; Palumbo, J.S.; Mullins, E.S.; Divanovic, S.; Weiler, H.; Owens, A.P.; Mackman, N.; Goss, A.; et al. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J. Clin. Investig. 2017, 127, 3152–3166. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, B.G.; Uygun, K.; Yarmush, M.L.; Saeidi, N. Surgical models of Roux-en-Y gastric bypass surgery and sleeve gastrectomy in rats and mice. Nat. Protoc. 2015, 10, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, P.; Chukwudi, C.; Pannu, P.R.; He, S.; Saeidi, N. Rewiring of the Liver Transcriptome across Multiple Time-Scales Is Associated with the Weight Loss-Independent Resolution of NAFLD Following RYGB. Metabolites 2022, 12, 318. https://doi.org/10.3390/metabo12040318
Lei P, Chukwudi C, Pannu PR, He S, Saeidi N. Rewiring of the Liver Transcriptome across Multiple Time-Scales Is Associated with the Weight Loss-Independent Resolution of NAFLD Following RYGB. Metabolites. 2022; 12(4):318. https://doi.org/10.3390/metabo12040318
Chicago/Turabian StyleLei, Peng, Chijioke Chukwudi, Prabh R. Pannu, Shijie He, and Nima Saeidi. 2022. "Rewiring of the Liver Transcriptome across Multiple Time-Scales Is Associated with the Weight Loss-Independent Resolution of NAFLD Following RYGB" Metabolites 12, no. 4: 318. https://doi.org/10.3390/metabo12040318
APA StyleLei, P., Chukwudi, C., Pannu, P. R., He, S., & Saeidi, N. (2022). Rewiring of the Liver Transcriptome across Multiple Time-Scales Is Associated with the Weight Loss-Independent Resolution of NAFLD Following RYGB. Metabolites, 12(4), 318. https://doi.org/10.3390/metabo12040318