
Emerging Role of Nuclear Receptors for the Treatment of NAFLD and NASH


 ,
,
Abstract
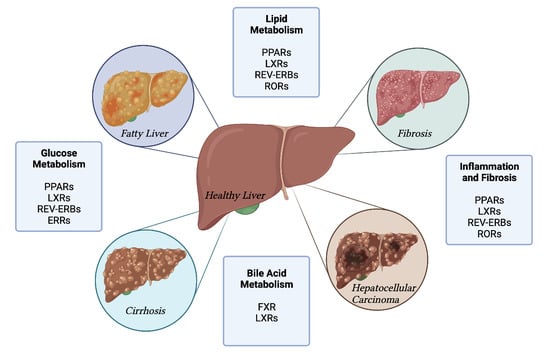
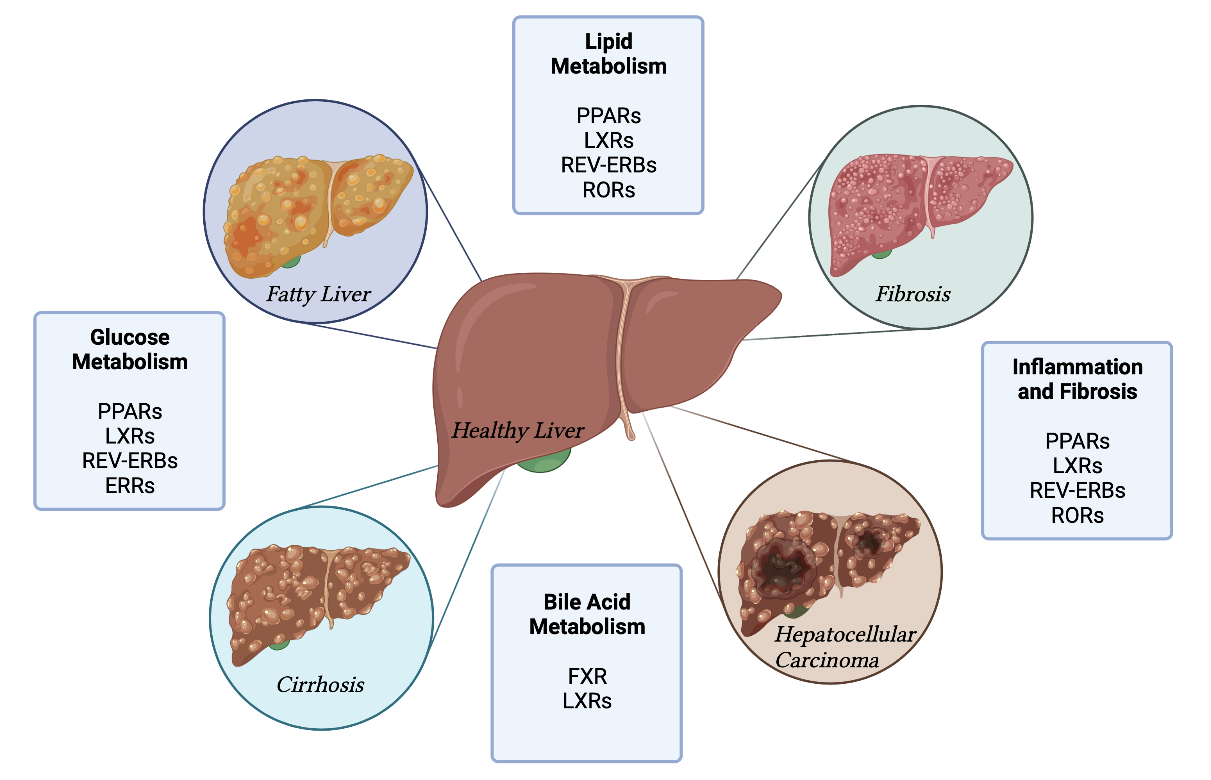
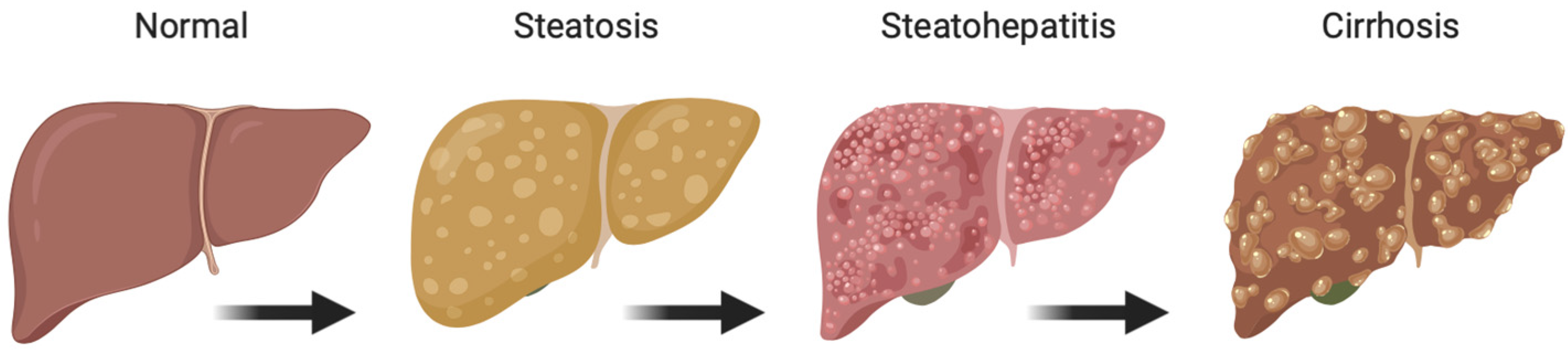
1. Introduction
2. Nuclear Receptor Targets for NAFLD
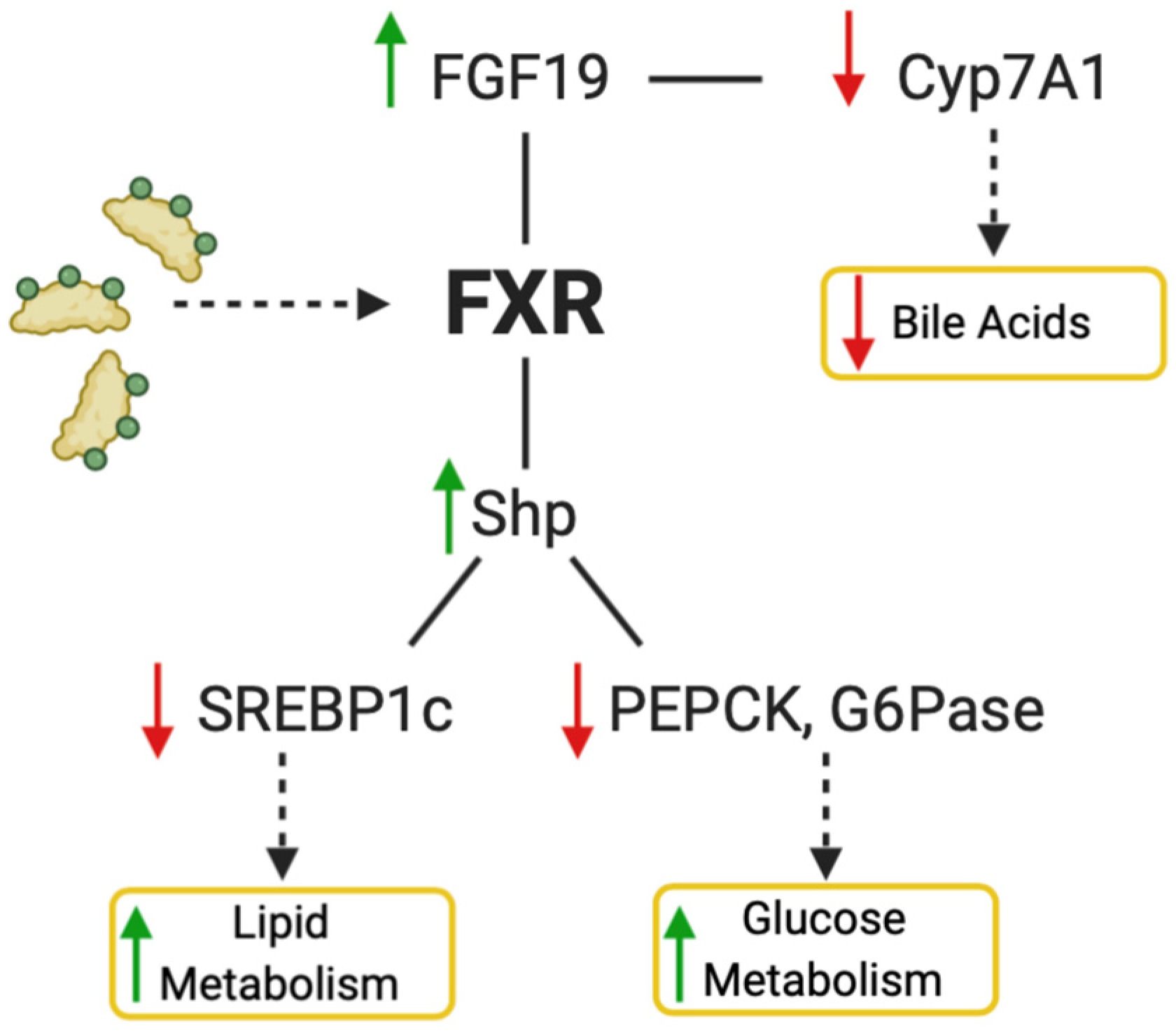
2.1. FXR
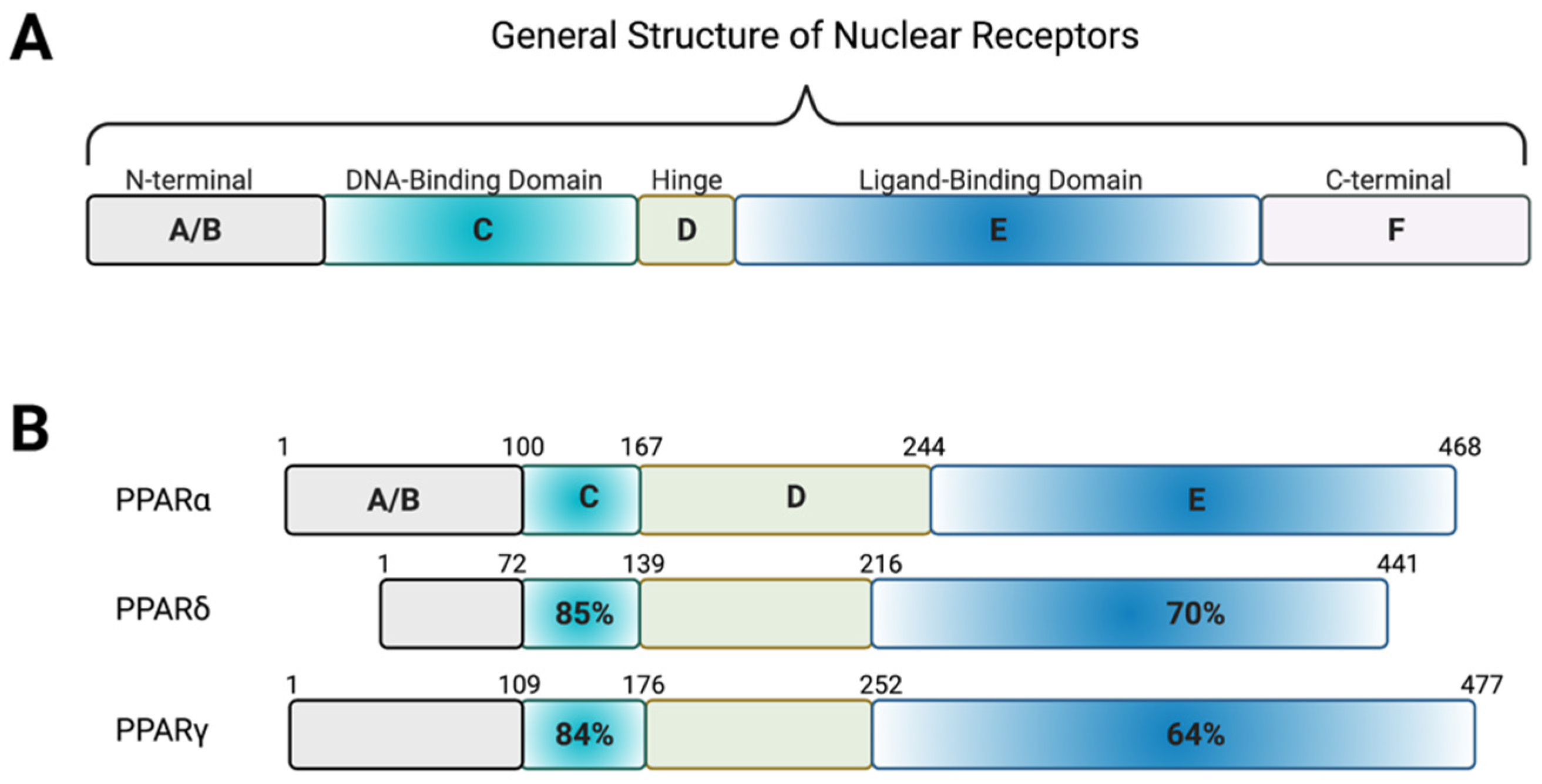
2.2. PPARs
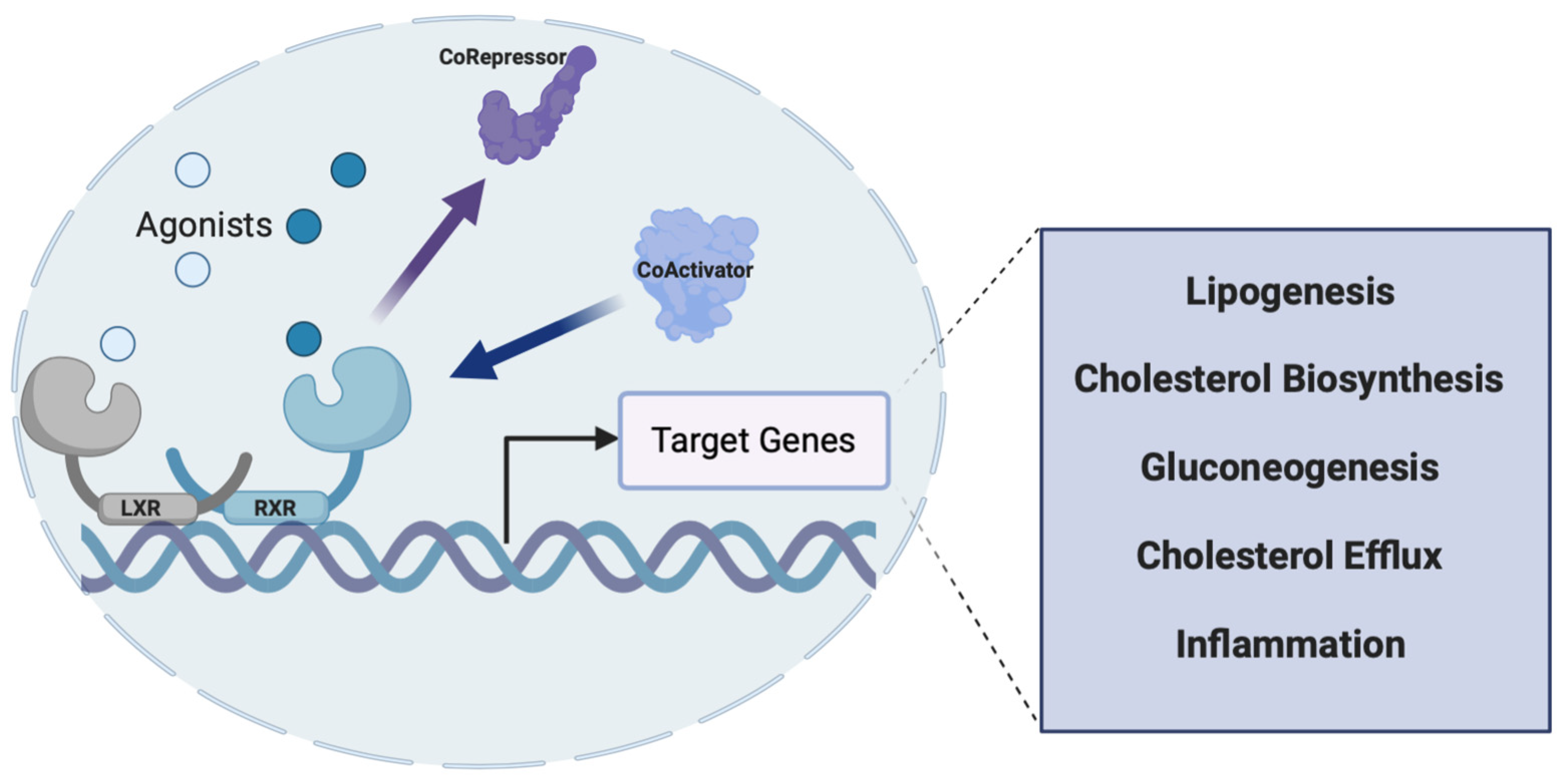
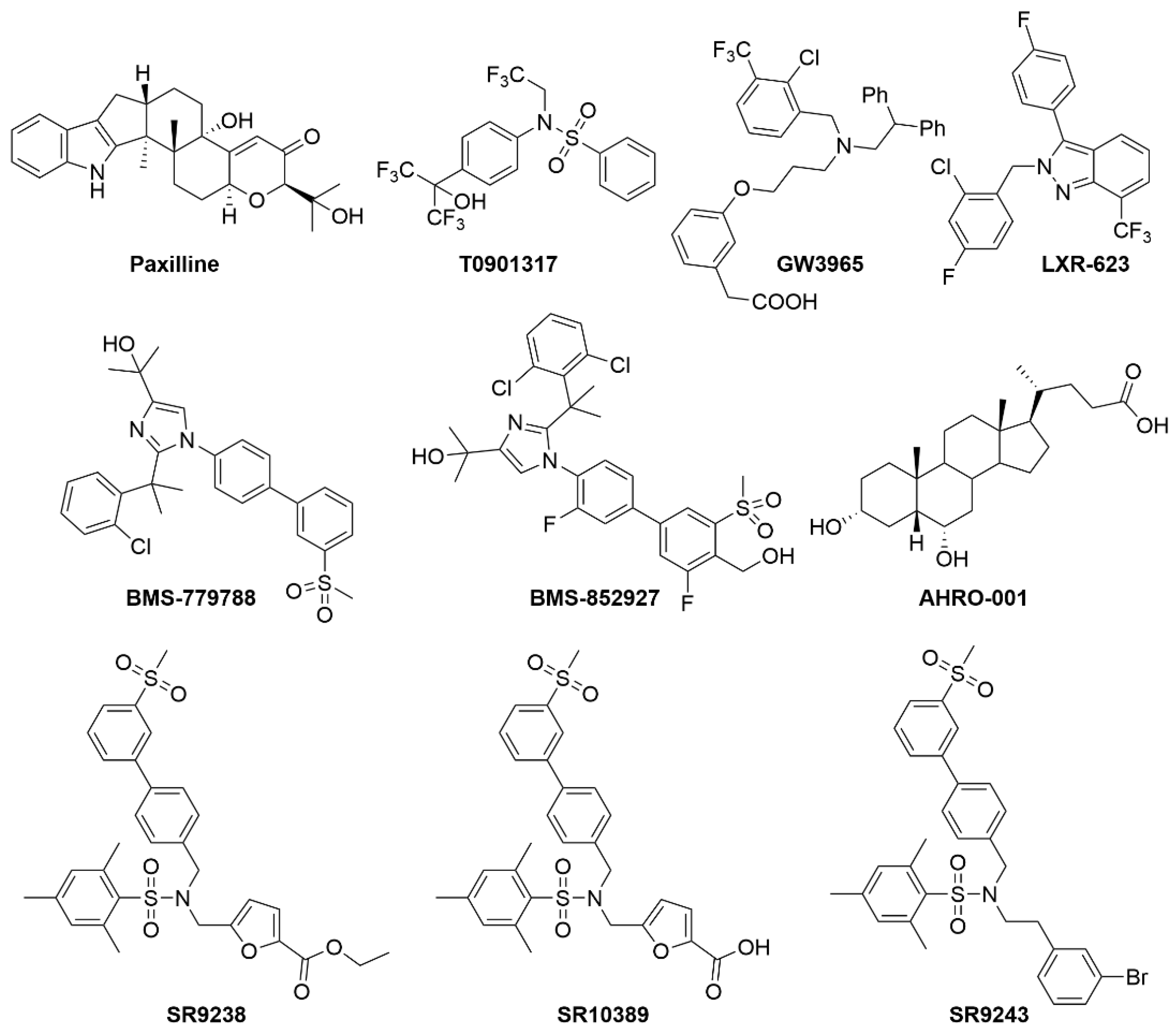
2.3. LXRs
2.4. RORs
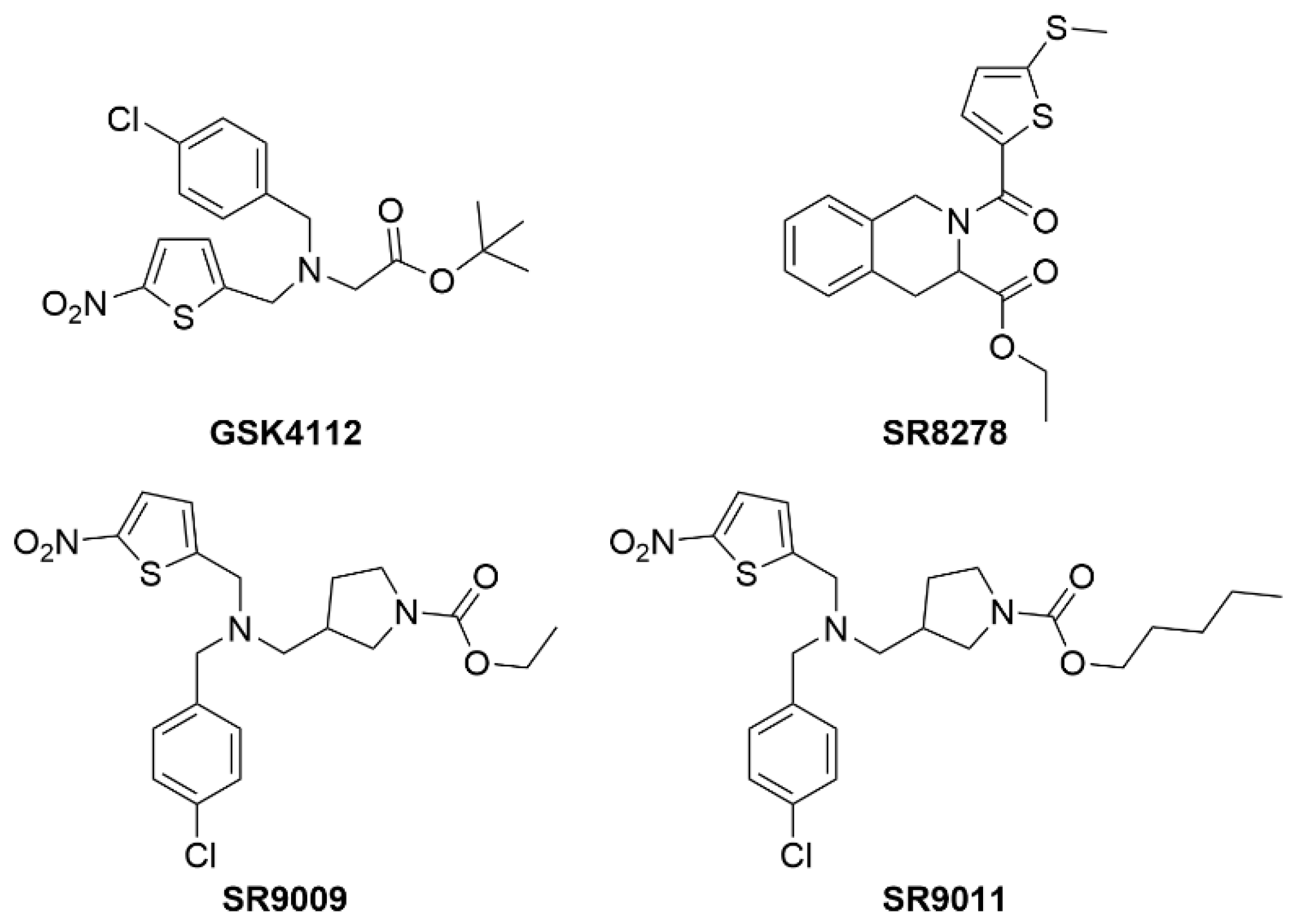
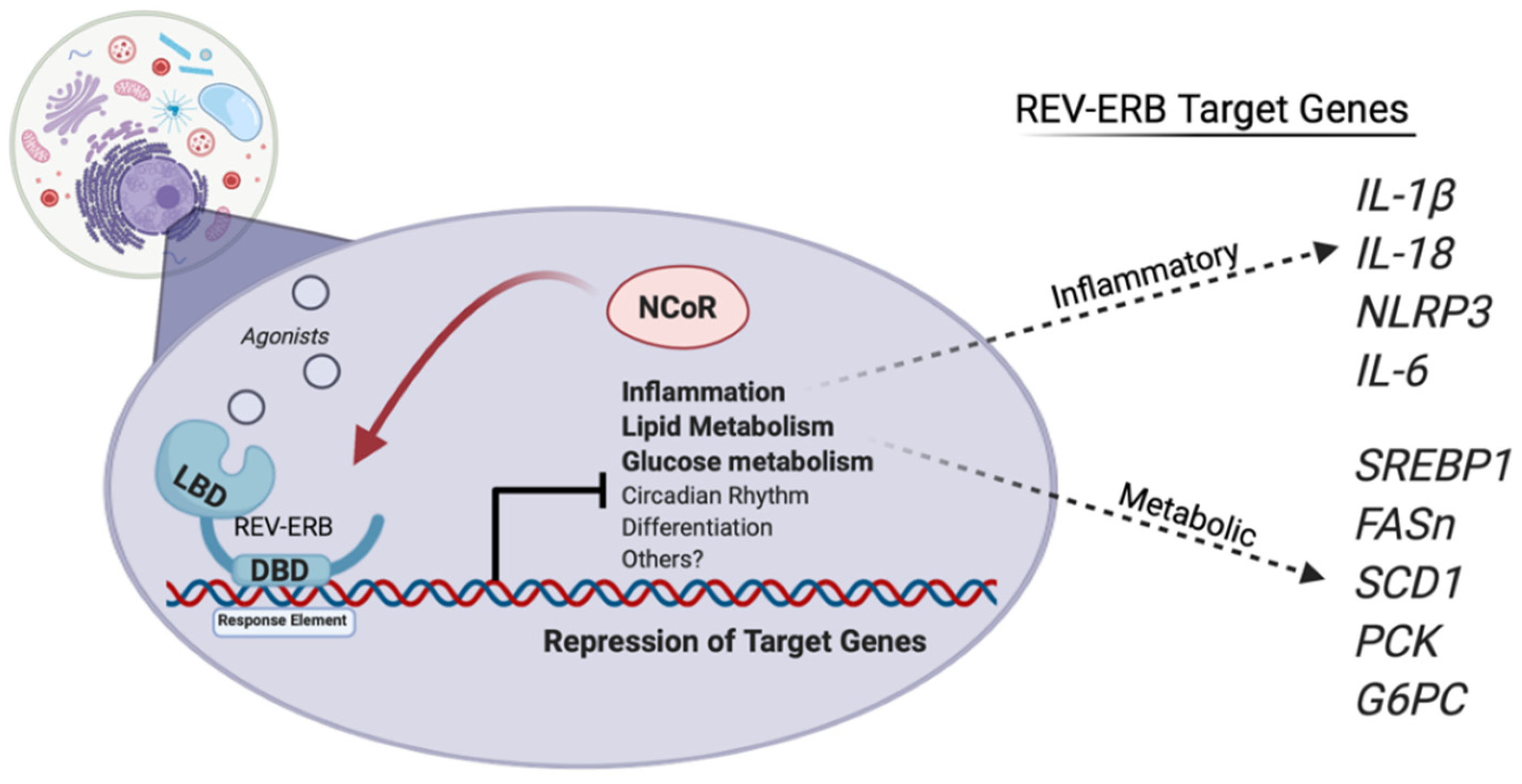
2.5. REV-ERBs
2.6. ERRs
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Bieghs, V.; Rensen, P.C.N.; Hofker, M.H.; Shiri-Sverdlov, R. NASH and atherosclerosis are two aspects of a shared disease: Central role for macrophages. Atherosclerosis 2012, 220, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Walenbergh, S.M.A.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol. 2013, 58, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Oligschlaeger, Y.; Shiri-Sverdlov, R. NAFLD Preclinical Models: More than a Handful, Less of a Concern? Biomedicines 2020, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chanda, D.; van Gorp, P.J.; Jeurissen, M.L.; Houben, T.; Walenbergh, S.M.; Debets, J.; Oligschlaeger, Y.; Gijbels, M.J.; Neumann, D.; et al. Macrophage Stimulating Protein Enhances Hepatic Inflammation in a NASH Model. PLoS ONE 2016, 11, e0163843. [Google Scholar] [CrossRef]
- Houben, T.; Penders, J.; Oligschlaeger, Y.; Dos Reis, I.A.M.; Bonder, M.J.; Koonen, D.P.; Fu, J.; Hofker, M.H.; Shiri-Sverdlov, R. Hematopoietic Npc1 mutation shifts gut microbiota composition in Ldlr−/− mice on a high-fat, high-cholesterol diet. Sci. Rep. 2019, 9, 14956. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S. Fatty liver: How frequent is it and why? Ann. Hepatol. 2004, 3, 63–65. [Google Scholar] [CrossRef]
- Liss, K.H.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef]
- Scaglioni, F.; Ciccia, S.; Marino, M.; Bedogni, G.; Bellentani, S. ASH and NASH. Dig. Dis. 2011, 29, 202–210. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P. Liver enzymes, nonalcoholic fatty liver disease, and incident cardiovascular disease. Hepatology 2010, 53, 375. [Google Scholar] [CrossRef] [PubMed]
- Targher, G. Obesity and Diabetes. Diabet. Med. 2006, 23, 1388. [Google Scholar] [CrossRef]
- Targher, G. Non-alcoholic fatty liver disease as a determinant of cardiovascular disease. Atherosclerosis 2007, 190, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Lonardo, A.; Targher, G. Is liver fat detrimental to vessels?: Intersections in the pathogenesis of NAFLD and atherosclerosis. Clin. Sci. 2008, 115, 1–12. [Google Scholar] [CrossRef]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Jiangao, F.S. The role of endotoxin, Kupffer cell and its related cytokines in the pathogenesis of nonalcoholic steatohepatitis in rats. Gastroenterology 2003, 124, A758–A759. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Kadayifçi, A. Nonalcoholic steatohepatitis: Role of leptin in pathogenesis and benefits of metformin in treatment. Am. J. Gastroenterol. 2003, 98, 2330. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Sakaida, I.; Okita, K. The role of oxidative stress in NASH and fatty liver model. Hepatol. Res. 2005, 33, 128–131. [Google Scholar] [CrossRef]
- van de Wier, B.; Haenen, G.R.M.M.; Koek, G.H.; Bast, A. Increase of oxidative stress in NASH by increased levels of citrate. Free Radic. Biol. Med. 2012, 53, S160. [Google Scholar] [CrossRef]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Shiota, G.; Tsuchiya, H. Pathophysiology of NASH: Insulin Resistance, Free Fatty Acids and Oxidative Stress. J. Clin. Biochem. Nutr. 2006, 38, 127–132. [Google Scholar] [CrossRef]
- Parola, M.; Novo, E. Nrf1 gene expression in the liver: A single gene linking oxidative stress to NAFLD, NASH and hepatic tumours. J. Hepatol. 2005, 43, 1096–1097. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Inzaugarat, M.E.; McGeough, M.D.; Holtmann, T.M.; Frissen, M.; Johnson, C.D.; Hoffman, H.H.; Feldstein, A.E.; Trautwein, C.; Wree, A. Direct activation of Nlrp3 inflammasome in hepatic stellate cells leads to upregulation of fibrotic markers. J. Hepatol. 2017, 66, S39. [Google Scholar] [CrossRef]
- Frissen, M.; Liao, L.; Bieghs, V.; Schneider, K.; Mohs, A.; Latz, E.; Wree, A.; Trautwein, C. Inability to form NLRP3 inflammasome complex leads to decreased inflammation and prevents fibrosis formation in mice after chronic bile duct ligation. Z. Gastroenterol. 2017, 56, E2–E89. [Google Scholar] [CrossRef]
- Szabo, G. Gut–Liver Axis in Alcoholic Liver Disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Horng, T.; Hotamisligil, G.S. Linking the inflammasome to obesity-related disease. Nat. Med. 2011, 17, 164–165. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. Klin. Wochenschr. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2013, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallee, D. Role of ER Stress in Inflammasome Activation and Non-Alcoholic Fatty Liver Disease Progression. Single Cell Biol. 2016, 5, 140. [Google Scholar] [CrossRef]
- Burris, T.P.; Solt, L.A.; Wang, Y.; Crumbley, C.; Banerjee, S.; Griffett, K.; Lundasen, T.; Hughes, T.; Kojetin, D.J. Nuclear Receptors and Their Selective Pharmacologic Modulators. Pharmacol. Rev. 2013, 65, 710–778. [Google Scholar] [CrossRef]
- Oro, A.E.; Hollenberg, S.M.; Evans, R.M. Transcriptional inhibition by a glucocorticoid receptor-β-galactosidase fusion protein. Cell 1988, 55, 1109–1114. [Google Scholar] [CrossRef]
- Jiao, Y.; Lu, Y.; Li, X. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2014, 36, 44–50. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Ma, K. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef]
- Cariou, B.; Duran-Sandoval, D.; Kuipers, F.; Staels, B. Farnesoid X Receptor: A New Player in Glucose Metabolism? Endocrinology 2005, 146, 981–983. [Google Scholar] [CrossRef]
- Yang, X.; Gonzalez, F.J.; Huang, M.; Bi, H. Nuclear receptors and non-alcoholic fatty liver disease: An update. Liver Res. 2020, 4, 88–93. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Role of FXR in Liver Inflammation During Nonalcoholic Steatohepatitis. Curr. Pharmacol. Rep. 2017, 3, 92–100. [Google Scholar] [CrossRef] [PubMed]
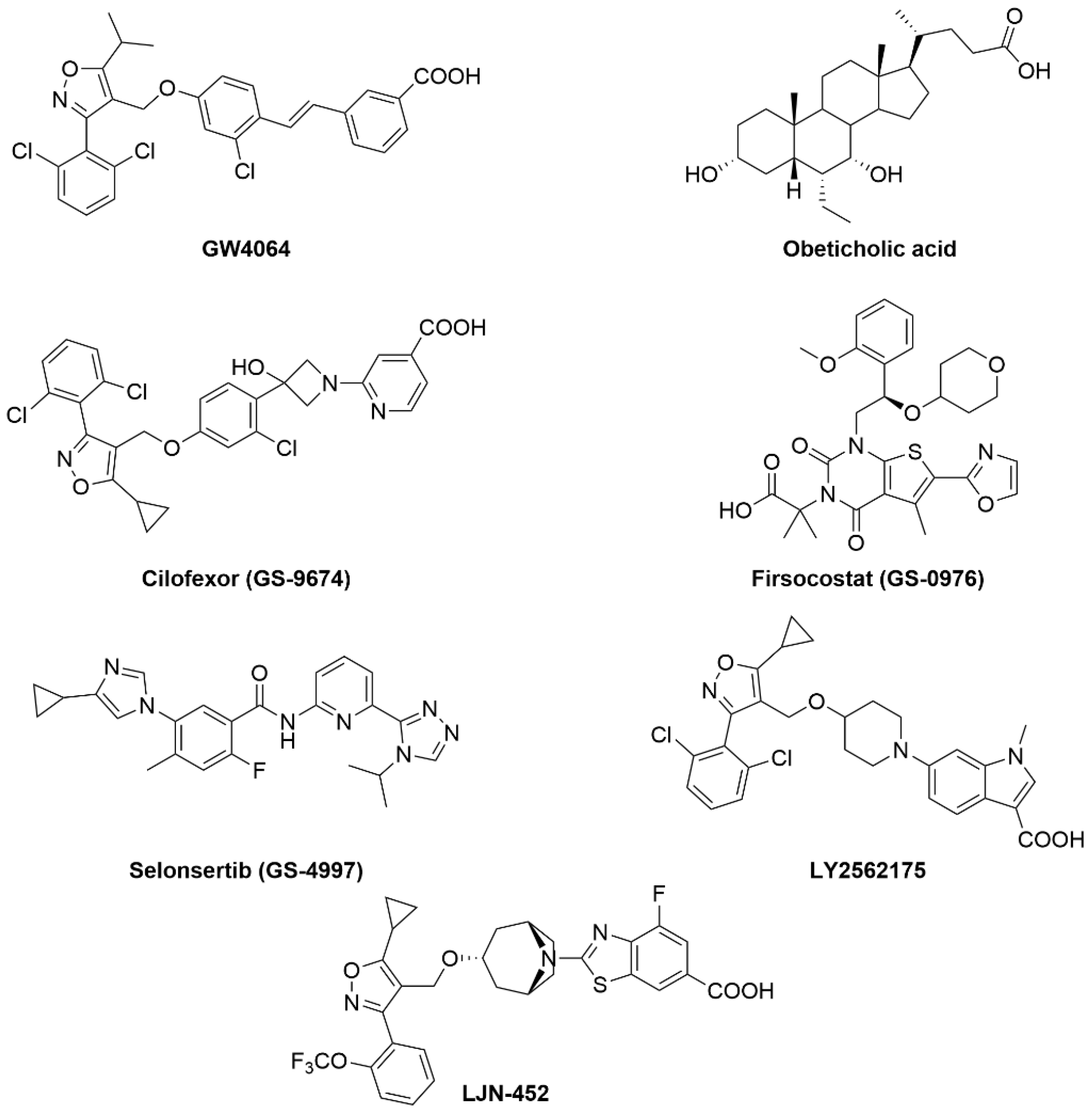
- Han, C. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Van Natta, M.L.; Tonascia, J.; Brunt, E.M.; Kleiner, D.E. Trials of obeticholic acid for non-alcoholic steatohepatitis—Authors’ reply. Lancet 2015, 386, 28–29. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Non-Cirrhotic Patients with Nonalcoholic Steatohepatitis: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Nelson, C.H.; Kirby, B.J.; Lu, N.; McColgan, B.; Djedjos, C.S.; Myers, R.P.; Cuvin, J.; Qin, A.; Mathias, A. Pharmacokinetics of selonsertib, GS-9674, and/or GS-0976 in combination in healthy subjects. J. Hepatol. 2017, 66, S151–S152. [Google Scholar] [CrossRef]
- Lawitz, E.; Gane, E.; Ruane, P.; Herring, R.; Younes, Z.H.; Kwo, P.; Zhang, J.; Jia, C.; Chuang, J.; McColgan, B.; et al. SAT-352-A combination of the ACC inhibitor GS-0976 and the nonsteroidal FXR agonist GS-9674 improves hepatic steatosis, biochemistry, and stiffness in patients with non-alcoholic steatohepatitis. J. Hepatol. 2019, 70, e794. [Google Scholar] [CrossRef]
- Lawitz, E.; Herring, R.; Younes, Z.H.; Gane, E.; Ruane, P.; Schall, R.A.; Jia, C.; Xu, R.; Mccolgan, B.; Djedjos, S.; et al. Su1522—Proof of Concept Study of an Apoptosis-Signal Regulating Kinase (ASK-1) Inhibitor (Selonsertib) in Combination with an Acetyl-Coa Carboxylase Inhibitor (GS-0976) or a Farnesoid X Receptor (FXR) Agonist (GS-9674) in NASH. Gastroenterology 2018, 154, S1166–S1167. [Google Scholar] [CrossRef]
- Alkhouri, N.; Lawitz, E.; Noureddin, M. Looking Into the Crystal Ball: Predicting the Future Challenges of Fibrotic NASH Treatment. Hepatol. Commun. 2019, 3, 605–613. [Google Scholar] [CrossRef]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef]
- Venetsanaki, V.; Karabouta, Z.; Polyzos, S.A. Farnesoid X nuclear receptor agonists for the treatment of nonalcoholic steatohepatitis. Eur. J. Pharmacol. 2019, 863, 172661. [Google Scholar] [CrossRef]
- Lin, J.H.; Zhang, J.J.; Lin, S.-L.; Chertow, G.M. Design of a Phase 2 Clinical Trial of an ASK1 Inhibitor, GS-4997, in Patients with Diabetic Kidney Disease. Nephron 2014, 129, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Tully, D.C.; Rucker, P.V.; Chianelli, D.; Williams, J.; Vidal, A.; Alper, P.B.; Mutnick, D.; Bursulaya, B.; Schmeits, J.; Wu, X.; et al. Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J. Med. Chem. 2017, 60, 9960–9973. [Google Scholar] [CrossRef] [PubMed]
- Genin, M.J.; Bueno, A.B.; Agejas Francisco, J.; Manninen, P.R.; Bocchinfuso, W.P.; Montrose-Rafizadeh, C.; Cannady, E.A.; Jones, T.M.; Stille, J.R.; Raddad, E.; et al. Discovery of 6-(4-{[5-Cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl]methoxy}piperidin-1-yl)-1-methyl-1H-indole-3-carboxylic Acid: A Novel FXR Agonist for the Treatment of Dyslipidemia. J. Med. Chem. 2015, 58, 9768–9772. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.; Lee, C.-H. PPARδ as a therapeutic target in metabolic disease. FEBS Lett. 2007, 582, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. PPARγ: A Nuclear Regulator of Metabolism, Differentiation, and Cell Growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Liu, Y.; Colby, J.; Zuo, X.; Jaoude, J.; Wei, D.; Shureiqi, I. The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int. J. Mol. Sci. 2018, 19, 3339. [Google Scholar] [CrossRef]
- Wolf, G. The Function of the Nuclear Receptor Peroxisome Proliferator–activated Receptor Delta in Energy Homeostasis. Nutr. Rev. 2003, 61, 387–390. [Google Scholar] [CrossRef][Green Version]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
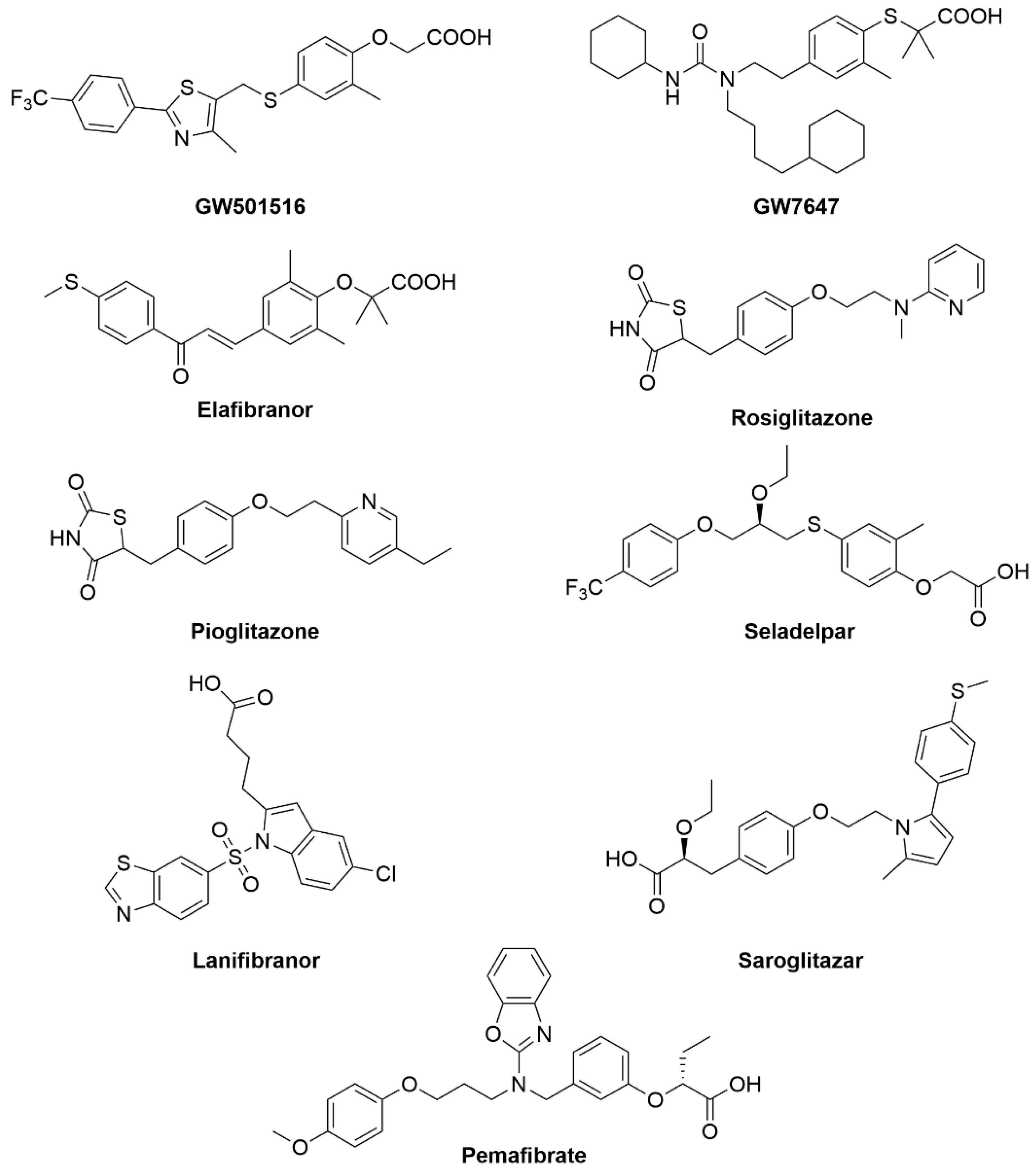
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator−Activated Receptor−α and −δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Therapeutic Landscape for NAFLD in 2020. Gastroenterology 2020, 158, 1984–1998.e3. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.P.; Caffrey, R.; Marioneaux, J.; Santhekadur, P.K.; Bhat, M.; Alonso, C.; Koduru, S.V.; Philip, B.; Jain, M.R.; Giri, S.R.; et al. The PPAR α/γ Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 9330. [Google Scholar] [CrossRef]
- Makled, M.N.; Sharawy, M.H.; El-Awady, M.S. The dual PPAR-α/γ agonist saroglitazar ameliorates thioacetamide-induced liver fibrosis in rats through regulating leptin. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2019, 392, 1569–1576. [Google Scholar] [CrossRef]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Ranvir, R.; Kadam, S.; Patel, H.; Swain, P.; et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2017, 38, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.A.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and Bile Acid Metabolism Are Impaired in Mice Lacking the Nuclear Oxysterol Receptor LXRα. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Peet, D.J.; Janowski, B.A.; Mangelsdorf, D.J. The LXRs: A new class of oxysterol receptors. Curr. Opin. Genet. Dev. 1998, 8, 571–575. [Google Scholar] [CrossRef]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXR and LXR. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D.; Tontonoz, P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis 2015, 242, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Tontonoz, P.; Mangelsdorf, D.J. Liver X Receptor Signaling Pathways in Cardiovascular Disease. Mol. Endocrinol. 2003, 17, 985–993. [Google Scholar] [CrossRef]
- Fu, X.; Menke, J.G.; Chen, Y.; Zhou, G.; MacNaul, K.L.; Wright, S.D.; Sparrow, C.P.; Lund, E.G. 27-Hydroxycholesterol Is an Endogenous Ligand for Liver X Receptor in Cholesterol-loaded Cells. J. Biol. Chem. 2001, 276, 38378–38387. [Google Scholar] [CrossRef]
- Brown, M.; Goldstein, J. Receptor-mediated control of cholesterol metabolism. Science 1976, 191, 150–154. [Google Scholar] [CrossRef]
- Gabbi, C.; Warner, M.; Gustafsson, J.-Å. Minireview: Liver X Receptor β: Emerging Roles in Physiology and Diseases. Mol. Endocrinol. 2009, 23, 129–136. [Google Scholar] [CrossRef]
- Gabbi, C.; Gustafsson, J.-Å. Bile acids in nonalcoholic steatohepatitis: Inserting nuclear receptors into the circle. Hepatology 2012, 56, 2008–2009. [Google Scholar] [CrossRef]
- Wang, M.; Thomas, J.; Burris, T.P.; Schkeryantz, J.; Michael, L.F. Molecular determinants of LXRα agonism. J. Mol. Graph. Model. 2003, 22, 173–181. [Google Scholar] [CrossRef]
- Bramlett, K.S.; Houck, K.A.; Borchert, K.M.; Dowless, M.S.; Kulanthaivel, P.; Zhang, Y.; Beyer, T.P.; Schmidt, R.; Thomas, J.S.; Michael, L.F.; et al. A Natural Product Ligand of the Oxysterol Receptor, Liver X Receptor. J. Pharmacol. Exp. Ther. 2003, 307, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qiu, D.K.; Ma, X. Liver X receptors bridge hepatic lipid metabolism and inflammation. J. Dig. Dis. 2012, 13, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- Castrillo, A.; Joseph, S.B.; Marathe, C.; Mangelsdorf, D.J.; Tontonoz, P. Liver X Receptor-dependent Repression of Matrix Metalloproteinase-9 Expression in Macrophages. J. Biol. Chem. 2003, 278, 10443–10449. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Walczak, R.; Dhamko, H.; Bradley, M.N.; Marathe, C.; Boyadjian, R.; Salazar, J.V.; Tontonoz, P. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: Identification of ARL7 as a direct target. J. Lipid Res. 2011, 52, 531–539. [Google Scholar] [CrossRef]
- Laffitte, B.A.; Repa, J.J.; Joseph, S.B.; Wilpitz, D.C.; Kast, H.R.; Mangelsdorf, D.J.; Tontonoz, P. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 507–512. [Google Scholar] [CrossRef]
- Bradley, M.N.; Tontonoz, P. Lesion Macrophages Are a Key Target for the Antiatherogenic Effects of LXR Agonists. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 10–11. [Google Scholar] [CrossRef]
- Calkin, A.C.; Tontonoz, P. Liver X Receptor Signaling Pathways and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1513–1518. [Google Scholar] [CrossRef]
- Joseph, S.B.; McKilligin, E.; Pei, L.; Watson, M.A.; Collins, A.R.; Laffitte, B.A.; Chen, M.; Noh, G.; Goodman, J.; Hagger, G.N.; et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7604–7609. [Google Scholar] [CrossRef]
- Terasaka, N.; Hiroshima, A.; Koieyama, T.; Ubukata, N.; Morikawa, Y.; Nakai, D.; Inaba, T. T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice. FEBS Lett. 2002, 536, 6–11. [Google Scholar] [CrossRef]
- Wójcik-Cichy, K.; Koślińska-Berkan, E.; Piekarska, A. The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clin. Exp. Hepatol. 2018, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Guleria, A. Patients with non-alcoholic fatty liver disease (NAFLD) have an increased risk of atherosclerosis and cardiovascular disease. Trop. Gastroenterol. 2013, 34, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Veca, V.; Gandolfo, V.; Natali, C.; Barsotti, F.; Lupattelli, G.; Siepi, D.; Ricci, M.; Vaudo, G. Pre-clinical vascular damage in metabolic syndrome: Correlation between nafld and carotid disease. Atherosclerosis 2018, 275, e191. [Google Scholar] [CrossRef]
- Pojskic, L.; Stimjanin, E.; Selimovic, H.; Pojskic, B. Risk factor for NAFLD and CHD—Similarity and differences. Atherosclerosis 2017, 263, e259. [Google Scholar] [CrossRef]
- VanWagner, L.B. New insights into NAFLD and subclinical coronary atherosclerosis. J. Hepatol. 2018, 68, 890–892. [Google Scholar] [CrossRef]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Mitro, N.; Vargas, L.; Romeo, R.; Koder, A.; Saez, E. T0901317 is a potent PXR ligand: Implications for the biology ascribed to LXR. FEBS Lett. 2007, 581, 1721–1726. [Google Scholar] [CrossRef]
- Kim, H.-H.; Seol, W.-G. T0901317 as an Inhibitor of Transcriptional Activation of Constitutive Androstane Receptor (CAR). J. Life Sci. 2011, 21, 481–485. [Google Scholar] [CrossRef]
- Chuu, C.P.; Chen, R.Y.; Hiipakka, R.A.; Kokontis, J.M.; Warner, K.V.; Xiang, J.; Liao, S. The liver X receptor agonist T0901317 acts as androgen receptor antagonist in human prostate cancer cells. Biochem. Biophys. Res. Commun. 2007, 357, 341–346. [Google Scholar] [CrossRef]
- Kanno, Y.; Tanuma, N.; Takahashi, A.; Inouye, Y. T0901317, a potent LXR agonist, is an inverse agonist of CAR. J. Toxicol. Sci. 2013, 38, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Leik, C.E.; Carson, N.L.; Hennan, J.K.; Basso, M.D.; Liu, Q.-Y.; Crandall, D.L.; Nambi, P. GW3965, a synthetic liver X receptor (LXR) agonist, reduces angiotensin II-mediated pressor responses in Sprague-Dawley rats. J. Cereb. Blood Flow Metab. 2007, 151, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Kotokorpi, P.; Ellis, E.; Parini, P.; Nilsson, L.-M.; Strom, S.; Steffensen, K.R.; Gustafsson, J.; Mode, A. Physiological Differences between Human and Rat Primary Hepatocytes in Response to Liver X Receptor Activation by 3-[3-[N-(2-Chloro-3-trifluoromethylbenzyl)-(2,2-diphenylethyl)amino]propyloxy]phenylacetic Acid Hydrochloride (GW3965). Mol. Pharmacol. 2007, 72, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Prawitt, J.; Beil, F.T.; Staels, B.; Heeren, J.; Niemeier, A. MS326 LXR activation with T0901317 and GW3965 in mice: Differential effects on liver and intestine, but no negative impact on bone. Atheroscler. Suppl. 2010, 11, 175. [Google Scholar] [CrossRef]
- Narce, M.; Poisson, J.-P. Lipid metabolism: Is liver X receptor (LXR) a regulator of adipocyte differentiation? Consequences of stearoyl-CoA desaturase activation by LXR. Curr. Opin. Lipidol. 2004, 15, 703–706. [Google Scholar] [CrossRef]
- Griesel, B.A.; Weems, J.; Russell, R.A.; Abel, E.D.; Humphries, K.; Olson, A.L. Acute Inhibition of Fatty Acid Import Inhibits GLUT4 Transcription in Adipose Tissue, but Not Skeletal or Cardiac Muscle Tissue, Partly Through Liver X Receptor (LXR) Signaling. Diabetes 2010, 59, 800–807. [Google Scholar] [CrossRef]
- Xiao, L.; Xie, X.; Zhai, Y. Functional crosstalk of CAR–LXR and ROR–LXR in drug metabolism and lipid metabolism. Adv. Drug Deliv. Rev. 2010, 62, 1316–1321. [Google Scholar] [CrossRef]
- Katz, A.; Udata, C.; Ott, E.; Hickey, L.; Burczynski, M.E.; Burghart, P.; Vesterqvist, O.; Meng, X. Safety, Pharmacokinetics, and Pharmacodynamics of Single Doses of LXR-623, a Novel Liver X-Receptor Agonist, in Healthy Participants. J. Clin. Pharmacol. 2009, 49, 643–649. [Google Scholar] [CrossRef]
- Griffett, K.; Solt, L.A.; El-Gendy, B.E.-D.M.; Kamenecka, T.M.; Burris, T.P. A Liver-Selective LXR Inverse Agonist That Suppresses Hepatic Steatosis. ACS Chem. Biol. 2012, 8, 559–567. [Google Scholar] [CrossRef]
- Griffett, K.; Welch, R.D.; Flaveny, C.A.; Kolar, G.R.; Neuschwander-Tetri, B.A.; Burris, T.P. The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol. Metab. 2015, 4, 353–357. [Google Scholar] [CrossRef]
- Flaveny, C.A.; Griffett, K.; El-Gendy, B.E.D.M.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M.; et al. Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Kumar, N.; He, Y.; Kamenecka, T.M.; Griffin, P.R.; Burris, T.P. Identification of a Selective RORγ Ligand That Suppresses TH17 Cells and Stimulates T Regulatory Cells. ACS Chem. Biol. 2012, 7, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V.; Tini, M.; Flock, G.; Ong, E.; Evans, R.M.; Otulakowski, G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994, 8, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z. Requirement for RORgamma in Thymocyte Survival and Lymphoid Organ Development. Science 2000, 288, 2369–2373. [Google Scholar] [CrossRef]
- Billon, C.; Sitaula, S.; Burris, T.P. Inhibition of RORα/γ suppresses atherosclerosis via inhibition of both cholesterol absorption and inflammation. Mol. Metab. 2016, 5, 997–1005. [Google Scholar] [CrossRef]
- André, E.; Gawlas, K.; Steinmayr, M.; Becker-André, M. A novel isoform of the orphan nuclear receptor RORβ is specifically expressed in pineal gland and retina. Gene 1998, 216, 277–283. [Google Scholar] [CrossRef]
- Medvedev, A.; Yan, Z.-H.; Hirose, T.; Giguère, V.; Jetten, A.M. Cloning of a cDNA encoding the murine orphan receptor RZR/RORγ and characterization of its response element. Gene 1996, 181, 199–206. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, N.; Solt, L.A.; Richardson, T.I.; Helvering, L.M.; Crumbley, C.; Garcia-Ordonez, R.D.; Stayrook, K.R.; Zhang, X.; Novick, S.; et al. Modulation of Retinoic Acid Receptor-related Orphan Receptor α and γ Activity by 7-Oxygenated Sterol Ligands. J. Biol. Chem. 2009, 285, 5013–5025. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, N.; Crumbley, C.; Griffin, P.R.; Burris, T.P. A second class of nuclear receptors for oxysterols: Regulation of RORα and RORγ activity by 24S-hydroxycholesterol (cerebrosterol). Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2010, 1801, 917–923. [Google Scholar] [CrossRef]
- Kumar, N.; Kojetin, D.J.; Solt, L.A.; Kumar, K.G.; Nuhant, P.; Duckett, D.R.; Cameron, M.D.; Butler, A.A.; Roush, W.R.; Griffin, P.R.; et al. Identification of SR3335 (ML-176): A Synthetic RORα Selective Inverse Agonist. ACS Chem. Biol. 2010, 6, 218–222. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, N.; Nuhant, P.; Cameron, M.D.; Istrate, M.A.; Roush, W.R.; Griffin, P.R.; Burris, T.P. Identification of SR1078, a Synthetic Agonist for the Orphan Nuclear Receptors RORα and RORγ. ACS Chem. Biol. 2010, 5, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Kumar, N.; Nuhant, P.; Wang, Y.; Lauer, J.L.; Liu, J.; Istrate, M.A.; Kamenecka, T.M.; Roush, W.R.; Vidović, D.; et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011, 472, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Hong, S.; Lee, M.; Jung, H.; Cho, W.-J.; Kim, E.-J.; Son, H.-Y.; Lee, M.-O.; Park, H.-G. N-methylthioureas as new agonists of retinoic acid receptor-related orphan receptor. Arch. Pharmacal Res. 2012, 35, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.R.; Leung, M.W.; Huang, P.; Ryan, D.A.; Krout, M.R.; Malapaka, R.R.; Chow, J.; Manel, N.; Ciofani, M.; Kim, S.V.; et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature 2011, 472, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Okamoto, K.; Takeda, Y.; Beak, J.Y.; Gerrish, K.; Bortner, C.D.; DeGraff, L.M.; Wada, T.; Xie, W.; Jetten, A.M. Transcriptional profiling reveals a role for RORα in regulating gene expression in obesity-associated inflammation and hepatic steatosis. Physiol. Genom. 2011, 43, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; Fitzsimmons, R.L.; Pearen, M.A.; Watt, M.J.; Muscat, G.E.O. Homozygous staggerer (sg/sg) mice display improved insulin sensitivity and enhanced glucose uptake in skeletal muscle. Diabetologia 2011, 54, 1169–1180. [Google Scholar] [CrossRef]
- Billon, C.; Sitaula, S.; Burris, T.P. Metabolic Characterization of a Novel RORα Knockout Mouse Model without Ataxia. Front. Endocrinol. 2017, 8, 141. [Google Scholar] [CrossRef]
- Kim, H.-J.; Han, Y.-H.; Na, H.; Kim, J.-Y.; Kim, T.; Kim, H.-J.; Shin, C.; Lee, J.W.; Lee, M.-O. Liver-specific deletion of RORα aggravates diet-induced nonalcoholic steatohepatitis by inducing mitochondrial dysfunction. Sci. Rep. 2017, 7, 16041. [Google Scholar] [CrossRef]
- Molinaro, A.; Caesar, R.; L’Homme, L.; Koh, A.; Ståhlman, M.; Staels, B.; Bäckhed, F. Liver-specific RORα deletion does not affect the metabolic susceptibility to western style diet feeding. Mol. Metab. 2019, 23, 82–87. [Google Scholar] [CrossRef]
- Han, Y.-H.; Kim, H.-J.; Na, H.; Nam, M.-W.; Kim, J.-Y.; Kim, J.-S.; Koo, S.-H.; Lee, M.-O. RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep. 2017, 20, 124–135. [Google Scholar] [CrossRef]
- Kim, E.J.; Yoon, Y.S.; Hong, S.; Son, H.Y.; Na, T.Y.; Lee, M.H.; Kang, H.J.; Park, J.; Cho, W.J.; Kim, S.G.; et al. Retinoic acid receptor-related orphan receptor α-induced activation of adenosine monophosphate-activated protein kinase results in attenuation of hepatic steatosis. Hepatology 2012, 55, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Starmann, J.; Fälth, M.; Spindelböck, W.; Lanz, K.L.; Lackner, C.; Zatloukal, K.; Trauner, M.; Sültmann, H. Gene Expression Profiling Unravels Cancer-Related Hepatic Molecular Signatures in Steatohepatitis but Not in Steatosis. PLoS ONE 2012, 7, e46584. [Google Scholar] [CrossRef] [PubMed]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.; McGilvray, I.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Cox, B.; Yaish, D.; Gross, D.; Rosenberg, N.; Amblard, F.; Shemuelian, Z.; Gefen, M.; Korach, A.; Tirosh, O.; et al. Agonist of RORA Attenuates Non-Alcoholic Fatty Liver Progression in Mice via Upregulation of microRNA 122. Gastroenterology 2020, 159, 999–1014. [Google Scholar] [CrossRef]
- Ma, X. Interleukin-17 Integrates Hepatic Steatosis and Inflammation in Nonalcoholic Fatty Liver Disease. Gastroenterology 2011, 140, S702. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Mridha, A.; Wree, A.; Robertson, A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Vivoli, E.; Piombanti, B.; Marra, F. Modulation of the NLRP3 inflammasome pathway mediates the anti-inflammatory action of indoleamine dioxygenase in experimental NASH. Dig. Liver Dis. 2018, 50, 16. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Fang, Z.-M.; Liu, W.-Q. NLRP3 inflammasome activation from Kupffer cells is involved in liver fibrosis of Schistosoma japonicum-infected mice via NF-κB. Parasites Vectors 2019, 12, 29. [Google Scholar] [CrossRef]
- Billon, C.; Murray, M.H.; Avdagic, A.; Burris, T.P. RORγ regulates the NLRP3 inflammasome. J. Biol. Chem. 2018, 294, 10–19. [Google Scholar] [CrossRef]
- Gibbs, J.E.; Blaikley, J.; Beesley, S.; Matthews, L.; Simpson, K.D.; Boyce, S.H.; Farrow, S.N.; Else, K.J.; Singh, D.; Ray, D.W.; et al. The nuclear receptor REV-ERB mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. USA 2011, 109, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Duez, H.; Staels, B. Rev-erb-α: An integrator of circadian rhythms and metabolism. J. Appl. Physiol. 2009, 107, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Bugge, A.; Feng, D.; Everett, L.J.; Briggs, E.R.; Mullican, S.E.; Wang, F.; Jager, J.; Lazar, M.A. Rev-erb and Rev-erb coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012, 26, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, B.M.; Zierath, J.R. Circadian rhythms and exercise—Re-setting the clock in metabolic disease. Nat. Rev. Endocrinol. 2019, 15, 197–206. [Google Scholar] [CrossRef]
- Li, T.; Lakner, A.M.; Ghosh, S.; Bonkovsky, H.L.; Schrum, L.W. 378 functional role of rev-erb in modulation of hepatic stellate cell transdifferentiation. J. Hepatol. 2012, 56, S152. [Google Scholar] [CrossRef]
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.W.; DiTacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012, 485, 123–127. [Google Scholar] [CrossRef]
- Raghuram, S.; Stayrook, K.R.; Huang, P.; Rogers, P.M.; Nosie, A.K.; McClure, D.B.; Burris, L.L.; Khorasanizadeh, S.; Burris, T.; Rastinejad, F. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBα and REV-ERBβ. Nat. Struct. Mol. Biol. 2007, 14, 1207–1213. [Google Scholar] [CrossRef]
- Burris, T.P. Nuclear Hormone Receptors for Heme: REV-ERBα and REV-ERBβ Are Ligand-Regulated Components of the Mammalian Clock. Mol. Endocrinol. 2008, 22, 1509–1520. [Google Scholar] [CrossRef]
- Grant, D.; Yin, L.; Collins, J.L.; Parks, D.J.; Orband-Miller, L.A.; Wisely, G.B.; Joshi, S.; Lazar, M.A.; Willson, T.M.; Zuercher, W.J. GSK4112, a Small Molecule Chemical Probe for the Cell Biology of the Nuclear Heme Receptor Rev-erbα. ACS Chem. Biol. 2010, 5, 925–932. [Google Scholar] [CrossRef]
- Kojetin, D.J.; Wang, Y.; Kamenecka, T.M.; Burris, T.P. Identification of SR8278, a Synthetic Antagonist of the Nuclear Heme Receptor REV-ERB. ACS Chem. Biol. 2010, 6, 131–134. [Google Scholar] [CrossRef]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Raspé, E.; Duez, H.; Mansén, A.; Fontaine, C.; Fiévet, C.; Fruchart, J.C.; Vennström, B.; Staels, B. Identification of Rev-erbα as a physiological repressor of apoC-III gene transcription. J. Lipid Res. 2002, 43, 2172–2179. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K. Dual apoC-II mimetic and apoC-III antagonist for hypertriglyceridaemia. Nat. Rev. Cardiol. 2020, 17, 201. [Google Scholar] [CrossRef] [PubMed]
- Anzulovich, A.; Mir, A.; Brewer, M.; Ferreyra, G.; Vinson, C.; Baler, R. Elovl3, a model gene to dissect homeostatic links between the circadian clock and nutritional status. J. Lipid Res. 2006, 47, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Le Martelot, G.; Claudel, T.; Gatfield, D.; Schaad, O.; Kornmann, B.; Sasso, G.L.; Moschetta, A.; Schibler, U. REV-ERBα Participates in Circadian SREBP Signaling and Bile Acid Homeostasis. PLoS Biol. 2009, 7, e1000181. [Google Scholar] [CrossRef] [PubMed]
- Claudel, T.; Duez, H.; van der Veen, J.; Fontaine, C.; Havinga, R.; Duhem, C.; Bloks, V.; Vennstr??m, B.; Fruchart, J.; Staels, B.; et al. Role of the nuclear orphan receptor Rev-erbá; in the regulation of bile acid synthesis. Eur. J. Gastroenterol. Hepatol. 2006, 18, A44. [Google Scholar] [CrossRef]
- Duez, H.; van der Veen, J.N.; Duhem, C.; Pourcet, B.; Touvier, T.; Fontaine, C.; Derudas, B.; Baugé, E.; Havinga, R.; Bloks, V.W.; et al. Regulation of Bile Acid Synthesis by the Nuclear Receptor Rev-erbα. Gastroenterology 2008, 135, 689–698.e5. [Google Scholar] [CrossRef]
- Chiang, J.Y. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef]
- Sitaula, S.; Billon, C.; Kamenecka, T.M.; Solt, L.A.; Burris, T.P. Suppression of atherosclerosis by synthetic REV-ERB agonist. Biochem. Biophys. Res. Commun. 2015, 460, 566–571. [Google Scholar] [CrossRef]
- Sitaula, S.; Zhang, J.; Ruiz, F.; Burris, T.P. Rev-erb regulation of cholesterologenesis. Biochem. Pharmacol. 2017, 131, 68–77. [Google Scholar] [CrossRef]
- Griffett, K.; Bedia-Diaz, G.; Elgendy, B.; Burris, T.P. REV-ERB agonism improves liver pathology in a mouse model of NASH. PLoS ONE 2020, 15, e0236000. [Google Scholar] [CrossRef] [PubMed]
- Caratti, G.; Iqbal, M.; Hunter, L.; Kim, D.; Wang, P.; Vonslow, R.M.; Begley, N.; Tetley, A.J.; Woodburn, J.L.; Pariollaud, M.; et al. REVERBa couples the circadian clock to hepatic glucocorticoid action. J. Clin. Am. Soc. Clin. Investig. 2018, 128, 4454–4471. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Pourcet, B.; Zecchin, M.; Ferri, L.; Beauchamp, J.; Sitaula, S.; Billon, C.; Delhaye, S.; Vanhoutte, J.; Mayeuf-Louchart, A.; Thorel, Q.; et al. Nuclear Receptor Subfamily 1 Group D Member 1 Regulates Circadian Activity of NLRP3 Inflammasome to Reduce the Severity of Fulminant Hepatitis in Mice. Gastroenterology 2018, 154, 1449–1464.e20. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lin, Y.; Yuan, X.; Li, F.; Guo, L.; Wu, B. REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. Nat. Commun. 2018, 9, 4246. [Google Scholar] [CrossRef]
- Reitz, C.J.; Alibhai, F.J.; Khatua, T.N.; Rasouli, M.; Bridle, B.W.; Burris, T.P.; Martino, T.A. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun. Biol. 2019, 2, 353. [Google Scholar] [CrossRef]
- Guo, D.K.; Zhu, Y.; Sun, H.Y.; Xu, X.Y.; Zhang, S.; Hao, Z.B.; Wang, G.H.; Mu, C.C.; Ren, H.G. Pharmacological activation of REV-ERBα represses LPS-induced microglial activation through the NF-κB pathway. Acta Pharmacol. Sin. 2018, 40, 26–34. [Google Scholar] [CrossRef]
- Griffin, P.; Dimitry, J.M.; Sheehan, P.W.; Lananna, B.V.; Guo, C.; Robinette, M.L.; Hayes, M.E.; Cedeño, M.R.; Nadarajah, C.J.; Ezerskiy, L.A.; et al. Circadian clock protein Rev-erbα regulates neuroinflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 5102–5107. [Google Scholar] [CrossRef]
- Giguère, V.; Yang, N.; Segui, P.; Evans, R.M. Identification of a new class of steroid hormone receptors. Nature 1988, 331, 91–94. [Google Scholar] [CrossRef]
- Giguère, V. Transcriptional Control of Energy Homeostasis by the Estrogen-Related Receptors. Endocr. Rev. 2008, 29, 677–696. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Q.; McDonald, T.; Davidoff, M.J.; Bailey, W.; Bai, C.; Liu, Q.; Caskey, C. Identification of two hERR2-related novel nuclear receptors utilizing bioinformatics and inverse PCR. Gene 1999, 228, 101–109. [Google Scholar] [CrossRef]
- Sladek, R.; Bader, J.A.; Giguère, V. The orphan nuclear receptor estrogen-related receptor alpha is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol. Cell. Biol. 1997, 17, 5400–5409. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, É.; Giguére, V. The multiple universes of estrogen-related receptor α and γ in metabolic control and related diseases. Acta Pharmacol. Sin. 2014, 36, 51–61. [Google Scholar] [CrossRef]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2014, 33, 49–54. [Google Scholar] [CrossRef]
- Narkar, V.A.; Fan, W.; Downes, M.; Yu, R.T.; Jonker, J.W.; Alaynick, W.A.; Banayo, E.; Karunasiri, M.S.; Lorca, S.; Evans, R.M. Exercise and PGC-1α-Independent Synchronization of Type I Muscle Metabolism and Vasculature by ERRγ. Cell Metab. 2011, 13, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Alaynick, W.A.; Kondo, R.P.; Xie, W.; He, W.; Dufour, C.R.; Downes, M.; Jonker, J.W.; Giles, W.; Naviaux, R.K.; Giguère, V.; et al. ERRγ Directs and Maintains the Transition to Oxidative Metabolism in the Postnatal Heart. Cell Metab. 2007, 6, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Busch, B.B.; Stevens, W.C.; Martin, R.; Ordentlich, P.; Zhou, S.; Sapp, D.W.; Horlick, R.A.; Mohan, R. Identification of a Selective Inverse Agonist for the Orphan Nuclear Receptor Estrogen-Related Receptor α. J. Med. Chem. 2004, 47, 5593–5596. [Google Scholar] [CrossRef]
- Patch, R.J.; Searle, L.L.; Kim, A.J.; De, D.; Zhu, X.; Askari, H.B.; O’Neill, J.C.; Abad, M.C.; Rentzeperis, D.; Liu, J.; et al. Identification of Diaryl Ether-Based Ligands for Estrogen-Related Receptor α as Potential Antidiabetic Agents. J. Med. Chem. 2011, 54, 788–808. [Google Scholar] [CrossRef]
- Patch, R.J.; Huang, H.; Patel, S.; Cheung, W.; Xu, G.; Zhao, B.-P.; Beauchamp, D.A.; Rentzeperis, D.; Geisler, J.G.; Askari, H.B.; et al. Indazole-based ligands for estrogen-related receptor α as potential anti-diabetic agents. Eur. J. Med. Chem. 2017, 138, 830–853. [Google Scholar] [CrossRef]
- Rangwala, S.M.; Wang, X.; Calvo, J.A.; Lindsley, L.; Zhang, Y.; Deyneko, G.; Beaulieu, V.; Gao, J.; Turner, G.; Markovits, J. Estrogen-related Receptor γ Is a Key Regulator of Muscle Mitochondrial Activity and Oxidative Capacity. J. Biol. Chem. 2010, 285, 22619–22629. [Google Scholar] [CrossRef]
- Kim, D.-K.; Ryu, D.; Koh, M.; Lee, M.-W.; Lim, D.; Kim, M.-J.; Kim, Y.-H.; Cho, W.-J.; Lee, C.-H.; Park, S.B.; et al. Orphan Nuclear Receptor Estrogen-Related Receptor γ (ERRγ) Is Key Regulator of Hepatic Gluconeogenesis. J. Biol. Chem. 2012, 287, 21628–21639. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sladek, R.; Bader, J.-A.; Matthyssen, A.; Rossant, J.; Giguère, V. Placental abnormalities in mouse embryos lacking the orphan nuclear receptor ERR-β. Nature 1997, 388, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, S.; Grasfeder, L.L.; Haeffele, C.L.; Lobenhofer, E.K.; Chu, T.-M.; Wolfinger, R.; Kazmin, D.; Koves, T.R.; Muoio, D.M.; Chang, C.-Y.; et al. Receptor-Selective Coactivators as Tools to Define the Biology of Specific Receptor-Coactivator Pairs. Mol. Cell 2006, 24, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Charest-Marcotte, A.; Dufour, C.R.; Wilson, B.J.; Tremblay, A.M.; Eichner, L.J.; Arlow, D.H.; Mootha, V.K.; Giguère, V. The homeobox protein Prox1 is a negative modulator of ERR/PGC-1 bioenergetic functions. Genes Dev. 2010, 24, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Chaveroux, C.; Eichner, L.J.; Dufour, C.R.; Shatnawi, A.; Khoutorsky, A.; Bourque, G.; Sonenberg, N.; Giguère, V. Molecular and Genetic Crosstalks between mTOR and ERRα Are Key Determinants of Rapamycin-Induced Nonalcoholic Fatty Liver. Cell Metab. 2013, 17, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Herzog, B.; Cardenas, J.; Hall, R.K.; Villena, J.A.; Budge, P.J.; Giguère, V.; Granner, D.K.; Kralli, A. Estrogen-related Receptor α Is a Repressor of Phosphoenolpyruvate Carboxykinase Gene Transcription. J. Biol. Chem. 2005, 281, 99–106. [Google Scholar] [CrossRef]
- Luo, J.; Sladek, R.; Carrier, J.; Bader, J.-A.; Richard, D.; Giguère, V. Reduced Fat Mass in Mice Lacking Orphan Nuclear Receptor Estrogen-Related Receptor α. Mol. Cell. Biol. 2003, 23, 7947–7956. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Solis-Muñoz, P.; Fernández-Moreira, D.; Grau, M.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. NADPH oxidase is implicated in the pathogenesis of oxidative phosphorylation dysfunction in mice fed a high-fat diet. Sci. Rep. 2016, 6, 23664. [Google Scholar] [CrossRef]
- B’Chir, W.; Dufour, C.R.; Ouellet, C.; Yan, M.; Tam, I.S.; Andrzejewski, S.; Xia, H.; Nabata, K.; St-Pierre, J.; Giguere, V. Divergent Role of Estrogen-Related Receptor α in Lipid- and Fasting-Induced Hepatic Steatosis in Mice. Endocrinology 2018, 159, 2153–2164. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FXR | Cilofexor (phase 2) | FXR agonist | ↓ Serum bile acids ↓ Hepatic Steatosis |
| Cilofexor + Firsocostat (phase 2) | FXR agonist + ACC inhibitor | ↓ Hepatic Steatosis ↓ Liver Stiffness ↓ ALT | |
| Cilofexor + Firsocostat + Selonsertib (phase 2) | FXR agonist + ACC inhibitor + ASK1 inhibitor | Currently ongoing and awaiting results | |
| TERN-101/LY2562175 (phase 2) | FXR agonist | ↓ LDL ↓ TGs ↑ HDL | |
| EDP-305 (phase 2a) | FXR agonist | ↓ Hepatic steatosis ↓ ALT Side effects including pruritus, headaches, and GI issues were reported | |
| GW4064 | FXR agonist | ↓ Hepatic steatosis ↓ Hyperglycemia Poor bioavailability | |
| Obeticholic Acid (phase 2) | FXR agonist | ↓ Hepatic inflammation ↓ Fibrosis Observed increases in LDL in some patients but can be co-treated with statins | |
| PPAR | Thiazolidinediones (TZDs), (FDA approved for diabetes; phase 2 for NASH) | PPARγ agonist | ↑ Insulin sensitivity ↑ Peripheral glucose clearance ↓ Hepatic steatosis ↓ FFA |
| Seladelpar (phase 2) | PPARδ agonist | ↓ ALT ↓ LDL Terminated due to increased liver damage | |
| Saroglitazar (phase 2 for NAFLD; phase 3 for NASH) | PPARα/δ agonist | ↓ Hepatic steatosis ↓ Liver enzymes Approved in India for use in NASH | |
| Lanifibranor (phase 3) | Pan-PPAR agonist | ↓ Hepatic inflammation | |
| GW501516 | PPARδ agonist | ↑ Insulin sensitivity ↓ Hepatic steatosis Induced cancer in preclinical models | |
| LXR | T0901317 | LXRα/β agonist | ↓ Cellular cholesterol ↑ Cholesterol Efflux ↑ Hepatic lipogenesis Initially in clinical trials for atherosclerosis but removed due to increased hepatic steatosis |
| GW3965 | LXRα/β agonist | ↑ Glucokinase expression ↓ Gluconeogenesis ↓ Inflammation ↑ Plasma and liver TGs | |
| LXR-632 (phase 1) | LXRα/β agonist | ↑ Anti-atherogenic properties Terminated post-phase 1 due to treatment-emergent adverse events | |
| CS-8080 (phase 1) | LXRα/β agonist | Clinical trials were terminated due to undisclosed reasons for these compounds. | |
| BMS-779788 (phase 1) | LXRα/β agonist | ||
| BMS-852927 (phase 1) | LXRα/β agonist | ||
| AHRO-001 (phase 1) | LXRα/β agonist | ↑ HDL ↑ Anti-atherogenic properties | |
| SR9238 | Liver-specific LXRα/β inverse agonist | ↓ Hepatic steatosis ↓ Hepatic inflammation | |
| SR9243 | LXRα/β inverse agonist | ↓ Hepatic steatosis ↓ Hepatic inflammation Targets Warburg effect in cancer cells | |
| ROR | SR1078 | RORα/γ agonist | ↑ FGF21 expression ↑ G6Pase expression |
| SR1001 | RORα/γ inverse agonist | ↓ Th17 cell-driven hepatic inflammation | |
| REV-ERB | GSK4112 | Rev-erbα/β agonist | No in vivo activity |
| SR8278 | Rev-erbα/β antagonist | Not tested in NAFLD but drives muscle regeneration and improves glucose regulation via increased osteocyte turnover | |
| SR9009 | Rev-erbα/β agonist | ↓ Plasma cholesterol ↓ Hepatic fibrosis ↑ Lean muscle mass ↓ Fat mass ↓ Activation and expression of NLRP3 inflammasome | |
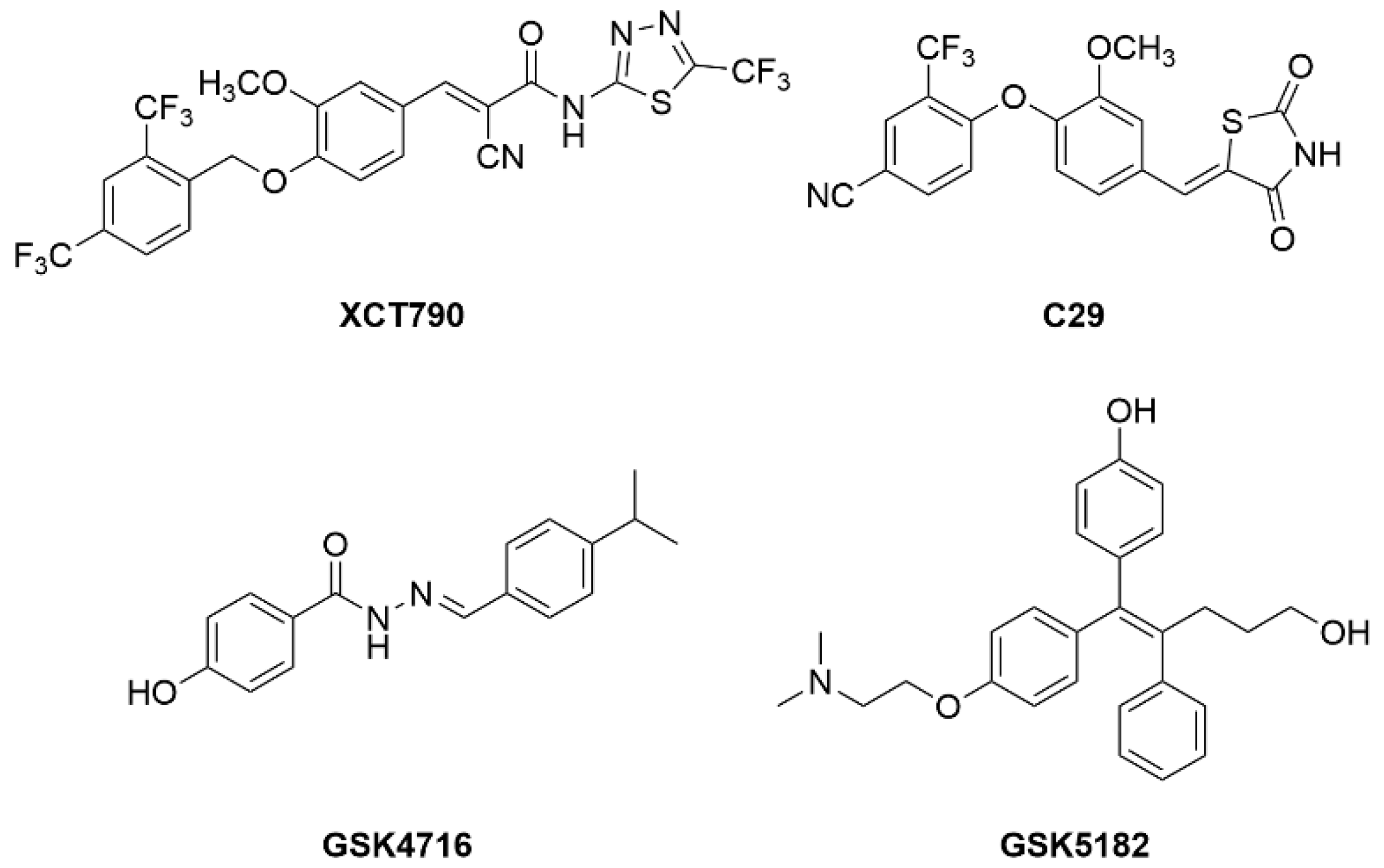
| ERR | XCT790 | ERRα inverse agonist | Anti-diabetic activity in rodents |
| GSK4716 | ERRβ/γ agonist | ↑ Mitochondrial function in myotubes | |
| GSK5182 | ERRγ inverse agonist | ↓ Plasma glucose in obese mice |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welch, R.D.; Billon, C.; Losby, M.; Bedia-Diaz, G.; Fang, Y.; Avdagic, A.; Elgendy, B.; Burris, T.P.; Griffett, K. Emerging Role of Nuclear Receptors for the Treatment of NAFLD and NASH. Metabolites 2022, 12, 238. https://doi.org/10.3390/metabo12030238
Welch RD, Billon C, Losby M, Bedia-Diaz G, Fang Y, Avdagic A, Elgendy B, Burris TP, Griffett K. Emerging Role of Nuclear Receptors for the Treatment of NAFLD and NASH. Metabolites. 2022; 12(3):238. https://doi.org/10.3390/metabo12030238
Chicago/Turabian StyleWelch, Ryan D., Cyrielle Billon, McKenna Losby, Gonzalo Bedia-Diaz, Yuanying Fang, Amer Avdagic, Bahaa Elgendy, Thomas P. Burris, and Kristine Griffett. 2022. "Emerging Role of Nuclear Receptors for the Treatment of NAFLD and NASH" Metabolites 12, no. 3: 238. https://doi.org/10.3390/metabo12030238
APA StyleWelch, R. D., Billon, C., Losby, M., Bedia-Diaz, G., Fang, Y., Avdagic, A., Elgendy, B., Burris, T. P., & Griffett, K. (2022). Emerging Role of Nuclear Receptors for the Treatment of NAFLD and NASH. Metabolites, 12(3), 238. https://doi.org/10.3390/metabo12030238