
The Potential of Nrf2 Activation as a Therapeutic Target in Systemic Lupus Erythematosus

Abstract
:1. Introduction
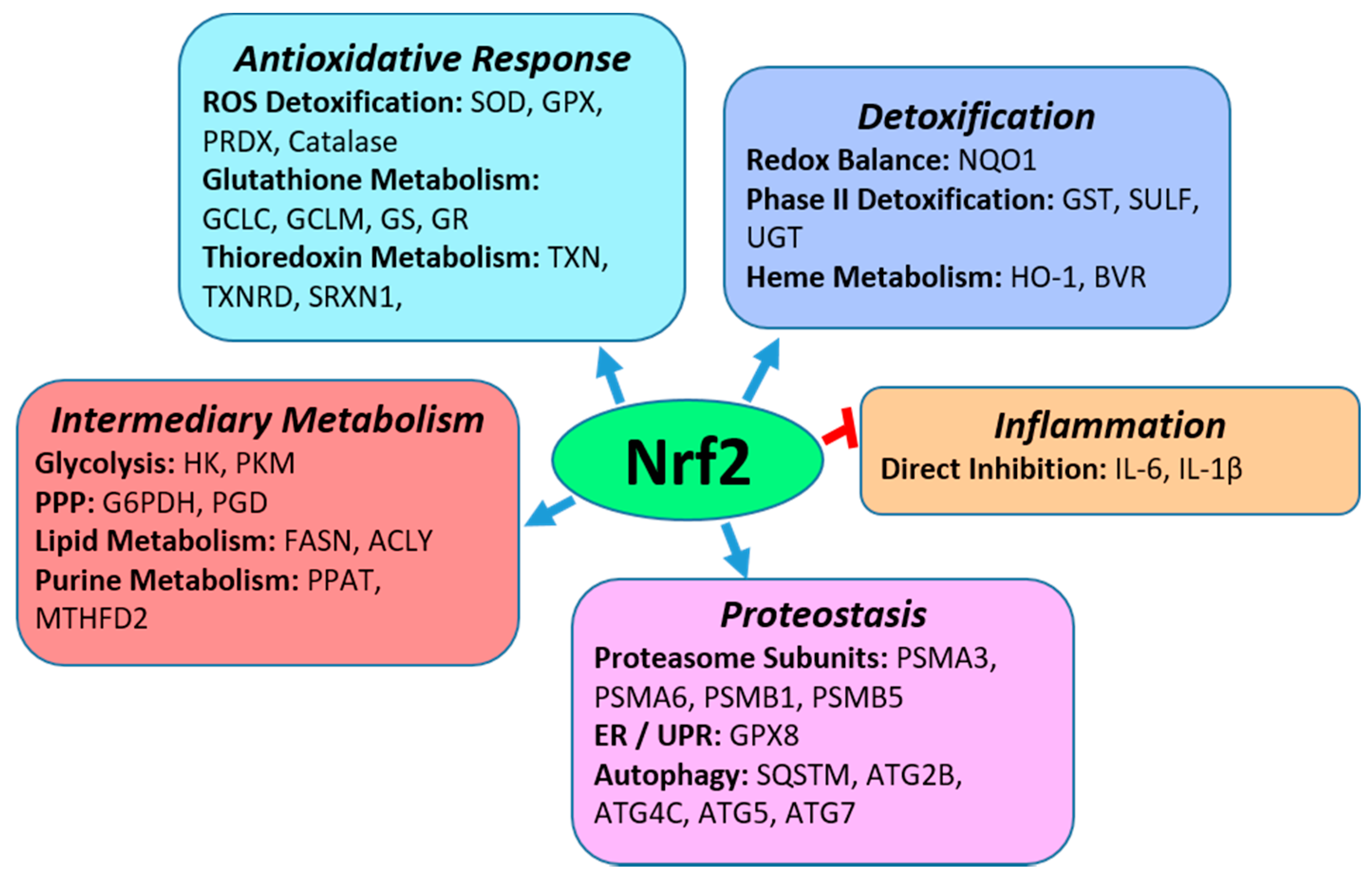
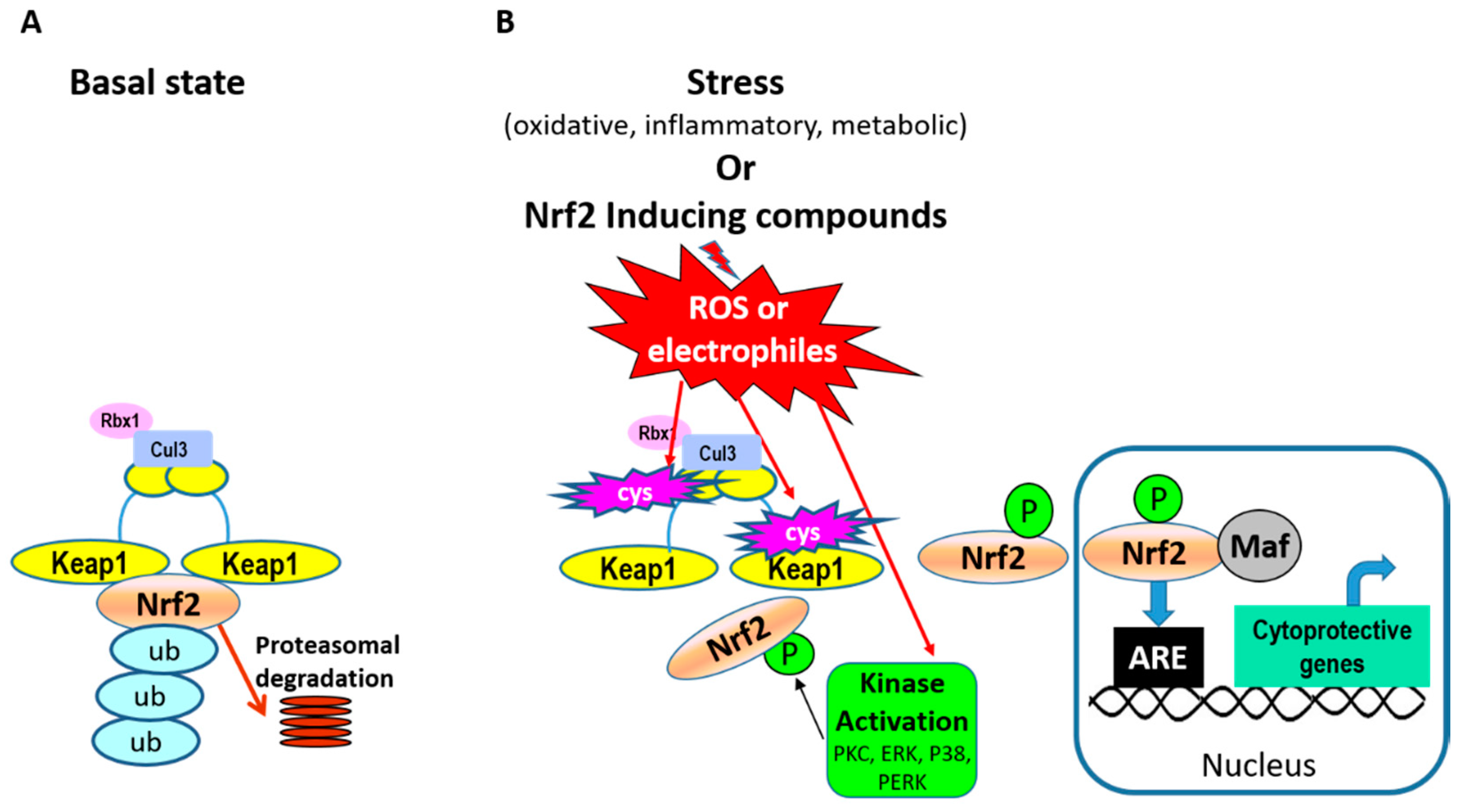
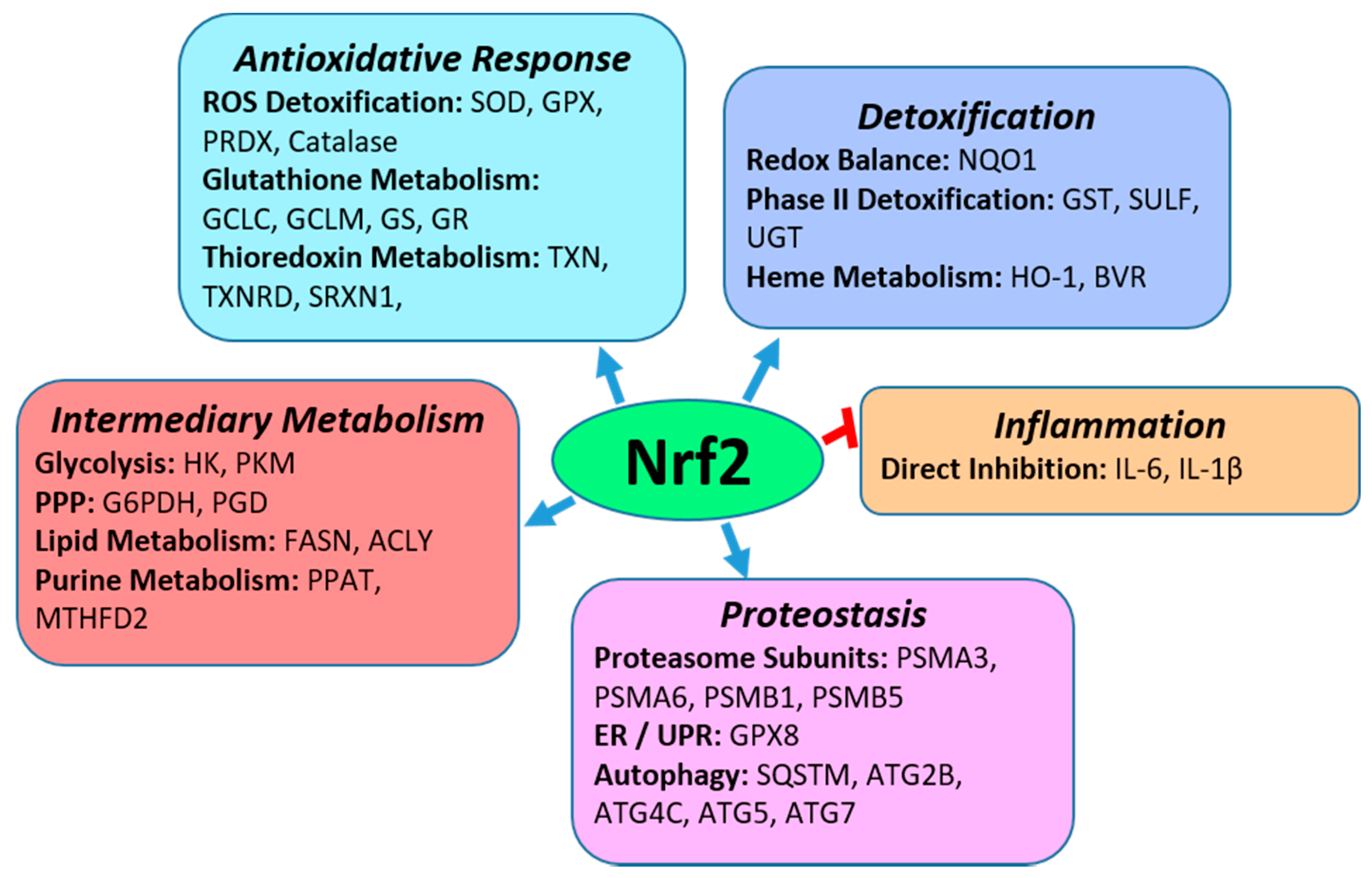
2. Nrf2
2.1. Nrf2 Regulation of Anti-Oxidative Responses and Cytoprotection
2.2. Nrf2 Regulation of Inflammation
3. Nrf2 in SLE and LN
3.1. Nrf2 in Animal Models of SLE and LN
3.1.1. Nrf2 Deficient Mice Develop Lupus-like Autoimmune Disease
3.1.2. Effects of Nrf2 Deficiency in Mouse Models of Spontaneous SLE-like Disease
3.1.3. Nrf2 is Regulated in Experimental Models of SLE
3.2. Nrf2 in Human SLE and LN
3.2.1. Nrf2 in Human Kidneys and Whole Blood
3.2.2. Nrf2 in Human Dendritic Cells
3.2.3. Nrf2 in Human T cells and NK Cells
3.2.4. Nrf2 and Antioxidative Gene Polymorphisms Associated with SLE
4. Nrf2 Inducers
4.1. Nrf2 Inducers in Animal Models of SLE
4.1.1. Sulforaphane
4.1.2. Dimethyl Fumarate
4.1.3. CDDO (2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oic Acid)
4.1.4. Epigallocatechin-3-Gallate (EGCG)
4.1.5. Artemisinin Derivatives
4.1.6. Baicalein
4.1.7. Dietary Extra Virgin Olive Oil (EVOO)
4.1.8. Oleuropein
4.1.9. Antroquinonol
4.1.10. Citral
4.2. Nrf2 Inducers in Studies of Cells from SLE Patients
4.2.1. Artesunate
4.2.2. Octyl Itaconate (OI)
4.3. Potential Utilization of Nrf2 Inducers for the Treatment of SLE in the Clinical Setting
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Bijl, M.; Reefman, E.; Horst, G.; Limburg, P.C.; Kallenberg, C.G. Reduced uptake of apoptotic cells by macrophages in systemic lupus erythematosus: Correlates with decreased serum levels of complement. Ann. Rheum. Dis. 2006, 65, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labonte, A.C.; Kegerreis, B.; Geraci, N.S.; Bachali, P.; Madamanchi, S.; Robl, R.; Catalina, M.D.; Lipsky, P.E.; Grammer, A.C. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS ONE 2018, 13, e0208132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A.; Burgos, P.I.; Cuitino, L.; Llanos, C. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 772. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Oxidative Stress in SLE T Cells, Is NRF2 Really the Target to Treat? Front. Immunol. 2021, 12, 633845. [Google Scholar] [CrossRef]
- Bomback, A.S.; Appel, G.B. Updates on the treatment of lupus nephritis. J. Am. Soc. Nephrol. 2010, 21, 2028–2035. [Google Scholar] [CrossRef]
- Davidson, J.E.; Fu, Q.; Ji, B.; Rao, S.; Roth, D.; Magder, L.S.; Petri, M. Renal Remission Status and Longterm Renal Survival in Patients with Lupus Nephritis: A Retrospective Cohort Analysis. J. Rheumatol. 2018, 45, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Perl, A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Bona, N.; Pezzarini, E.; Balbi, B.; Daniele, S.M.; Rossi, M.F.; Monje, A.L.; Basiglio, C.L.; Pelusa, H.F.; Arriaga, S.M.M. Oxidative stress, inflammation and disease activity biomarkers in lupus nephropathy. Lupus 2020, 29, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienhofer, D.; Boeltz, S.; Hoffmann, M.H. Reactive oxygen homeostasis - the balance for preventing autoimmunity. Lupus 2016, 25, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ma, J.; Deng, Y.; Kelly, J.A.; Kim, K.; Bang, S.Y.; Lee, H.S.; Li, Q.Z.; Wakeland, E.K.; Qiu, R.; et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat. Genet. 2017, 49, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001, 60, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Otsuki, A.; Suzuki, M.; Katsuoka, F.; Tsuchida, K.; Suda, H.; Morita, M.; Shimizu, R.; Yamamoto, M. Unique cistrome defined as CsMBE is strictly required for Nrf2-sMaf heterodimer function in cytoprotection. Free Radic. Biol. Med. 2016, 91, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS ONE 2009, 4, e6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Xu, C.; Pan, Z.; Keum, Y.S.; Kim, J.H.; Shen, G.; Yu, S.; Oo, K.T.; Ma, J.; Kong, A.N. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850. [Google Scholar] [CrossRef]
- Zhu, H.; Itoh, K.; Yamamoto, M.; Zweier, J.L.; Li, Y. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: Protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett. 2005, 579, 3029–3036. [Google Scholar] [CrossRef] [Green Version]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.; Zhang, M.; Yin, F. Heme Oxygenase-1, Oxidation, Inflammation, and Atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Choi, A.M. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Chanas, S.A.; Jiang, Q.; McMahon, M.; McWalter, G.K.; McLellan, L.I.; Elcombe, C.R.; Henderson, C.J.; Wolf, C.R.; Moffat, G.J.; Itoh, K.; et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 2002, 365, 405–416. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Banning, A.; Brigelius-Flohe, R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid. Redox Signal. 2005, 7, 889–899. [Google Scholar] [CrossRef]
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Lee, E.W.; Song, J.; Kim, H.R.; Jun, Y.C.; Hwang, K.A. MafK positively regulates NF-kappaB activity by enhancing CBP-mediated p65 acetylation. Sci. Rep. 2013, 3, 3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. et Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Lee, H.N.; Jang, J.H.; Kim, S.H.; Lee, Y.H.; Hahn, Y.I.; Ngo, H.K.; Choi, Y.; Joe, Y.; Chung, H.T.; et al. 15-Deoxy-Delta(12,14)-Prostaglandin J2 Exerts Proresolving Effects Through Nuclear Factor E2-Related Factor 2-Induced Expression of CD36 and Heme Oxygenase-1. Antioxid. Redox Signal. 2017, 27, 1412–1431. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [Green Version]
- Izui, S.; Kelley, V.E.; Masuda, K.; Yoshida, H.; Roths, J.B.; Murphy, E.D. Induction of various autoantibodies by mutant gene lpr in several strains of mice. J. Immunol. 1984, 133, 227–233. [Google Scholar]
- Han, S.; Zhuang, H.; Lee, P.Y.; Li, M.; Yang, L.; Nigrovic, P.A.; Reeves, W.H. NF-E2-Related Factor 2 Regulates Interferon Receptor Expression and Alters Macrophage Polarization in Lupus. Arthritis Rheumatol. 2020, 72, 1707–1720. [Google Scholar] [CrossRef]
- Klemm, P.; Rajendiran, A.; Fragoulis, A.; Wruck, C.; Schippers, A.; Wagner, N.; Bopp, T.; Tenbrock, K.; Ohl, K. Nrf2 expression driven by Foxp3 specific deletion of Keap1 results in loss of immune tolerance in mice. Eur. J. Immunol. 2020, 50, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Morzadec, C.; Macoch, M.; Sparfel, L.; Kerdine-Romer, S.; Fardel, O.; Vernhet, L. Nrf2 expression and activity in human T lymphocytes: Stimulation by T cell receptor activation and priming by inorganic arsenic and tert-butylhydroquinone. Free Radic. Biol. Med. 2014, 71, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.J.; Velardi, E.; Shono, Y.; Argyropoulos, K.V.; Holland, A.M.; Smith, O.M.; Yim, N.L.; Rao, U.K.; Kreines, F.M.; Lieberman, S.R.; et al. Nrf2 regulates CD4(+) T cell-induced acute graft-versus-host disease in mice. Blood 2018, 132, 2763–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, S.; Martina, M.N.; Bandapalle, S.; Racusen, L.C.; Potteti, H.R.; Hamad, A.R.; Reddy, S.P.; Rabb, H. T Lymphocyte-Specific Activation of Nrf2 Protects from AKI. J. Am. Soc. Nephrol. 2015, 26, 2989–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Murakami, S.; Biswal, S.S.; Sakaguchi, S.; Harigae, H.; Yamamoto, M.; Motohashi, H. Systemic Activation of NRF2 Alleviates Lethal Autoimmune Inflammation in Scurfy Mice. Mol. Cell. Biol. 2017, 37, e00063-17. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Fragoulis, A.; Klemm, P.; Baumeister, J.; Klock, W.; Verjans, E.; Boll, S.; Mollmann, J.; Lehrke, M.; Costa, I.; et al. Nrf2 Is a Central Regulator of Metabolic Reprogramming of Myeloid-Derived Suppressor Cells in Steady State and Sepsis. Front. Immunol. 2018, 9, 1552. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Stein, T.D.; Johnson, J.A. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol. Genom. 2004, 18, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Flemming, A.; Brummer, T.; Reth, M.; Jumaa, H. The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat. Immunol. 2003, 4, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.A.; Ding, W.Z.; Mohseni, M.; Treadwell, E.L.; Dooley, M.A.; St Clair, E.W.; Gilkeson, G.S.; Cooper, G.S. Glutathione S-transferase M null homozygosity and risk of systemic lupus erythematosus associated with sun exposure: A possible gene-environment interaction for autoimmunity. J. Rheumatol. 2003, 30, 276–282. [Google Scholar]
- Watanabe-Fukunaga, R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; Nagata, S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992, 356, 314–317. [Google Scholar] [CrossRef]
- Morito, N.; Yoh, K.; Hirayama, A.; Itoh, K.; Nose, M.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2 deficiency improves autoimmune nephritis caused by the fas mutation lpr. Kidney Int. 2004, 65, 1703–1713. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Chen, H.; Ding, Q.; Xu, X.; Yu, B.; Huang, Z. Nuclear Factor Erythroid 2-related Factor 2 Deficiency Exacerbates Lupus Nephritis in B6/lpr mice by Regulating Th17 Cell Function. Sci. Rep. 2016, 6, 38619. [Google Scholar] [CrossRef] [Green Version]
- Kelley, V.E.; Roths, J.B. Interaction of mutant lpr gene with background strain influences renal disease. Clin. Immunol. Immunopathol. 1985, 37, 220–229. [Google Scholar] [CrossRef]
- Satoh, M.; Kumar, A.; Kanwar, Y.S.; Reeves, W.H. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc. Natl. Acad. Sci. USA 1995, 92, 10934–10938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castejon, M.L.; Sánchez-Hidalgo, M.; Aparicio-Soto, M.; Montoya, T.; Martín-LaCave, I.; Fernández-Bolaños, J.G.; Alarcón-de-la-Lastra, C. Dietary oleuropein and its new acyl-derivate attenuate murine lupus nephritis through HO-1/Nrf2 activation and suppressing JAK/STAT, NF-κB, MAPK and NLRP3 inflammasome signaling pathways. J. Nutr. Biochem. 2019, 74, 108229. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shi, G.; Wang, J.; Zhang, D.; Pan, Y.; Dou, H.; Hou, Y. Baicalein ameliorates pristane-induced lupus nephritis via activating Nrf2/HO-1 in myeloid-derived suppressor cells. Arthritis Res. Ther. 2019, 21, 105. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Soto, M.; Sanchez-Hidalgo, M.; Cardeno, A.; Rosillo, M.A.; Sanchez-Fidalgo, S.; Utrilla, J.; Martin-Lacave, I.; Alarcon-de-la-Lastra, C. Dietary extra virgin olive oil attenuates kidney injury in pristane-induced SLE model via activation of HO-1/Nrf-2 antioxidant pathway and suppression of JAK/STAT, NF-kappaB and MAPK activation. J. Nutr. Biochem. 2016, 27, 278–288. [Google Scholar] [CrossRef]
- Ebihara, S.; Tajima, H.; Ono, M. Nuclear factor erythroid 2-related factor 2 is a critical target for the treatment of glucocorticoid-resistant lupus nephritis. Arthritis Res. Ther. 2016, 18, 139. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Qi, J.; Wang, J.; Pan, Y.; Li, J.; Xia, X.; Dou, H.; Hou, Y. Protective effect of dihydroartemisinin in inhibiting senescence of myeloid-derived suppressor cells from lupus mice via Nrf2/HO-1 pathway. Free Radic. Biol. Med. 2019, 143, 260–274. [Google Scholar] [CrossRef]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Lai, J.H.; Dai, M.S.; Jheng, H.L.; Kuo, M.T.; Chen, P.; Chen, A. Antroquinonol differentially modulates T cell activity and reduces interleukin-18 production, but enhances Nrf2 activation, in murine accelerated severe lupus nephritis. Arthritis Rheum. 2012, 64, 232–242. [Google Scholar] [CrossRef]
- Ka, S.M.; Lin, J.C.; Lin, T.J.; Liu, F.C.; Chao, L.K.; Ho, C.L.; Yeh, L.T.; Sytwu, H.K.; Hua, K.F.; Chen, A. Citral alleviates an accelerated and severe lupus nephritis model by inhibiting the activation signal of NLRP3 inflammasome and enhancing Nrf2 activation. Arthritis Res. Ther. 2015, 17, 331. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Chen, H.C.; Shui, H.A.; Li, C.Y.; Hua, K.F.; Chang, W.L.; Huang, J.J.; Yang, S.S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, Z.; Liu, H.; Jia, L.; Qin, M.; Wang, X. Exacerbating lupus nephritis following BPA exposure is associated with abnormal autophagy in MRL/lpr mice. Am. J. Transl. Res. 2020, 12, 649–659. [Google Scholar] [PubMed]
- Banerjee, N.; Wang, H.; Wang, G.; Boor, P.J.; Khan, M.F. Redox-sensitive Nrf2 and MAPK signaling pathways contribute to trichloroethene-mediated autoimmune disease progression. Toxicology 2021, 457, 152804. [Google Scholar] [CrossRef] [PubMed]
- Ene, C.D.; Georgescu, S.R.; Tampa, M.; Matei, C.; Mitran, C.I.; Mitran, M.I.; Penescu, M.N.; Nicolae, I. Cellular Response against Oxidative Stress, a Novel Insight into Lupus Nephritis Pathogenesis. J. Pers. Med. 2021, 11, 693. [Google Scholar] [CrossRef]
- Gautam, P.; Kaur, G.; Tandon, A.; Sharma, A.; Bhatnagar, A. Altered redox regulation by Nrf2-Keap1 system in dendritic cells of systemic lupus erythematosus patients. Lupus 2020, 29, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Tandon, A.; Anupam, K.; Kaushal, J.; Gautam, P.; Sharma, A.; Bhatnagar, A. Altered oxidative stress markers in relation to T cells, NK cells & killer immunoglobulin receptors that are associated with disease activity in SLE patients. Lupus 2020, 29, 1831–1844. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.A.A.; Mallmann, N.H.; de Souza, G.; de Jesus Bacha, T.; Lima, E.S.; de Lima, D.S.N.; de Souza Passos, L.F.; de Souza Gonçalves, M.; de Moura Neto, J.P. Glutathione S-transferase, catalase, and mitochondrial superoxide dismutase gene polymorphisms modulate redox potential in systemic lupus erythematosus patients from Manaus, Amazonas, Brazil. Clin. Rheumatol. 2021, 40, 3639–3649. [Google Scholar] [CrossRef] [PubMed]
- Warchoł, T.; Lianeri, M.; Wudarski, M.; Łacki, J.K.; Jagodziński, P.P. Catalase -262C>T polymorphism in systemic lupus erythematosus in Poland. Rheumatol. Int. 2008, 28, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Kim-Howard, X.; Sun, C.; Molineros, J.E.; Maiti, A.K.; Chandru, H.; Adler, A.; Wiley, G.B.; Kaufman, K.M.; Kottyan, L.; Guthridge, J.M.; et al. Allelic heterogeneity in NCF2 associated with systemic lupus erythematosus (SLE) susceptibility across four ethnic populations. Hum. Mol. Genet. 2014, 23, 1656–1668. [Google Scholar] [CrossRef] [Green Version]
- Córdova, E.J.; Velázquez-Cruz, R.; Centeno, F.; Baca, V.; Orozco, L. The NRF2 gene variant, -653G/A, is associated with nephritis in childhood-onset systemic lupus erythematosus. Lupus 2010, 19, 1237–1242. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yoh, K.; Kobayashi, A.; Ishii, Y.; Kure, S.; Koyama, A.; Sakamoto, T.; Sekizawa, K.; Motohashi, H.; Yamamoto, M. Identification of polymorphisms in the promoter region of the human NRF2 gene. Biochem. Biophys. Res. Commun. 2004, 321, 72–79. [Google Scholar] [CrossRef]
- Martini, S.; Nair, V.; Keller, B.J.; Eichinger, F.; Hawkins, J.J.; Randolph, A.; Böger, C.A.; Gadegbeku, C.A.; Fox, C.S.; Cohen, C.D.; et al. Integrative biology identifies shared transcriptional networks in CKD. J. Am. Soc. Nephrol. 2014, 25, 2559–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [Green Version]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef] [Green Version]
- Inoyama, D.; Chen, Y.; Huang, X.; Beamer, L.J.; Kong, A.N.; Hu, L. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J. Biomol. Screen 2012, 17, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Bader, S.; Wilmers, J.; Pelzer, M.; Jendrossek, V.; Rudner, J. Activation of anti-oxidant Keap1/Nrf2 pathway modulates efficacy of dihydroartemisinin-based monotherapy and combinatory therapy with ionizing radiation. Free Radic. Biol. Med. 2021, 168, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, J.A.; van Elven, E.H.; Hogendoorn, P.C.; Corver, W.E.; Hoedemaeker, P.J.; Fleuren, G.J. Murine chronic graft-versus-host disease as a model for lupus nephritis. Am. J. Pathol. 1988, 130, 639–641. [Google Scholar] [PubMed]
- Wu, X.; Zhang, W.; Shi, X.; An, P.; Sun, W.; Wang, Z. Therapeutic effect of artemisinin on lupus nephritis mice and its mechanisms. Acta Biochim. Biophys. Sin. 2010, 42, 916–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, O.; Zhang, H.; Gu, Z.; Zhao, S.; Xu, T.; Zhou, K.; Jiang, B.; Wang, J.; Zeng, X.; Sun, L. A pilot study of the therapeutic efficacy and mechanism of artesunate in the MRL/lpr murine model of systemic lupus erythematosus. Cell Mol. Immunol. 2009, 6, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; He, S.; Bai, B.; Zhang, L.; Xue, L.; Lin, Z.; Yang, X.; Zhu, F.; He, P.; Tang, W.; et al. Therapeutic effects of the artemisinin analog SM934 on lupus-prone MRL/lpr mice via inhibition of TLR-triggered B-cell activation and plasma cell formation. Cell. Mol. Immunol. 2016, 13, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, W.Z.; Li, H.; Jiang, B.; Nandakumar, K.S.; Liu, K.F.; Liu, L.X.; Yu, X.C.; Tan, H.J.; Zhou, C. Therapeutic effects of artesunate on lupus-prone MRL/lpr mice are dependent on T follicular helper cell differentiation and activation of JAK2-STAT3 signaling pathway. Phytomedicine 2019, 62, 152965. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane: Translational research from laboratory bench to clinic. Nutr. Rev. 2013, 71, 709–726. [Google Scholar] [CrossRef]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: An overview. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [Green Version]
- Liby, K.T.; Sporn, M.B. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharm. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Kanlaya, R.; Khamchun, S.; Kapincharanon, C.; Thongboonkerd, V. Protective effect of epigallocatechin-3-gallate (EGCG) via Nrf2 pathway against oxalate-induced epithelial mesenchymal transition (EMT) of renal tubular cells. Sci. Rep. 2016, 6, 30233. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Oesch, F. The immunosuppressive activity of artemisinin-type drugs towards inflammatory and autoimmune diseases. Med. Res. Rev. 2021, 41, 3023–3061. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Liu, Y.; Chen, L.; Cao, S.; Huang, Y.; Yang, X.; Zhu, F.; Tang, W.; He, S.; Zuo, J. Artemisinin analogue SM934 protects against lupus-associated antiphospholipid syndrome via activation of Nrf2 and its targets. Sci. China Life Sci. 2021, 64, 1702–1719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qi, S.; Song, Y.; Ling, C. Artemisinin attenuates early renal damage on diabetic nephropathy rats through suppressing TGF-beta1 regulator and activating the Nrf2 signaling pathway. Life Sci. 2020, 256, 117966. [Google Scholar] [CrossRef]
- Su, X.; Guo, W.; Yuan, B.; Wang, D.; Liu, L.; Wu, X.; Zhang, Y.; Kong, X.; Lin, N. Artesunate attenuates bone erosion in rheumatoid arthritis by suppressing reactive oxygen species via activating p62/Nrf2 signaling. Biomed. Pharm. 2021, 137, 111382. [Google Scholar] [CrossRef]
- Li, W.D.; Dong, Y.J.; Tu, Y.Y.; Lin, Z.B. Dihydroarteannuin ameliorates lupus symptom of BXSB mice by inhibiting production of TNF-alpha and blocking the signaling pathway NF-kappa B translocation. Int. Immunopharmacol. 2006, 6, 1243–1250. [Google Scholar] [CrossRef]
- Hou, L.F.; He, S.J.; Li, X.; Yang, Y.; He, P.L.; Zhou, Y.; Zhu, F.H.; Yang, Y.F.; Li, Y.; Tang, W.; et al. Oral administration of artemisinin analog SM934 ameliorates lupus syndromes in MRL/lpr mice by inhibiting Th1 and Th17 cell responses. Arthritis Rheum. 2011, 63, 2445–2455. [Google Scholar] [CrossRef]
- Feng, X.; Chen, W.; Xiao, L.; Gu, F.; Huang, J.; Tsao, B.P.; Sun, L. Artesunate inhibits type I interferon-induced production of macrophage migration inhibitory factor in patients with systemic lupus erythematosus. Lupus 2017, 26, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Wang, X.; Xie, Y.; Cai, X.; Yu, N.; Hu, Y.; Zheng, Z. 4-Octyl Itaconate Activates Nrf2 Signaling to Inhibit Pro-Inflammatory Cytokine Production in Peripheral Blood Mononuclear Cells of Systemic Lupus Erythematosus Patients. Cell. Physiol. Biochem. 2018, 51, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.; Adamczyk, A.; Kellerer, C.; Belge, K.; Bruck, J.; Berner, T.; Merten, K.; Nunez Gomez, N.; Neureither, M.; Rocken, M.; et al. Fumaderm(R) in daily practice for psoriasis: Dosing, efficacy and quality of life. Br. J. Derm. 2014, 171, 1197–1205. [Google Scholar] [CrossRef]
- Tsianakas, A.; Herzog, S.; Landmann, A.; Patsinakidis, N.; Perusquia Ortiz, A.M.; Bonsmann, G.; Luger, T.A.; Kuhn, A. Successful treatment of discoid lupus erythematosus with fumaric acid esters. J. Am. Acad. Derm. 2014, 71, e15–e17. [Google Scholar] [CrossRef]
- Saracino, A.M.; Orteu, C.H. Severe recalcitrant cutaneous manifestations in systemic lupus erythematosus successfully treated with fumaric acid esters. Br. J. Derm. 2017, 176, 472–480. [Google Scholar] [CrossRef]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants 2020, 9, 716. [Google Scholar] [CrossRef]
- To, C.; Ringelberg, C.S.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Sporn, M.B.; Liby, K.T. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 2015, 36, 769–781. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Nrf2−/− | ||||
|---|---|---|---|---|
| Strain | Age (Weeks) | Sex | Effects in Nrf2−/− Mice | Refs. |
| ICR | 25 | M and F |  kidney glomerular lesions kidney glomerular lesions | Yoh et al. [15] |
| 25 | M and F | serum IgG, anti dsDNA | ||
| 25 | M and F | creatinine clearance | ||
| 50 | F | ↑ spleen/body weight ratio; | ||
| germinal center hyperplasia | ||||
| 60 | F | ↑serum IgG, anti dsDNA | ||
| 60 | F | ↑ kidney glomerular lesions | ||
| 60 | F | ↓ creatinine clearance | ||
| 60 | F | ↑ lipid peroxidation in subcutaneous fat | ||
| 60 | F | ↓ CD19-CD3+ and CD4+CD8- lymphocytes | ||
| 100 | F | none survived | ||
| 15; 25; 50; 70; 100 | F | survival rate (%): 100; 100; 75–80; 60; 0 | ||
| 15; 25; 50; 70; 100 | M | survival rate (%): 100; 80; 65; 60; 20–25 | ||
| C57B6/129SVJ | 20 | F | some kidney glomerular IgG, IgM, C3 | Li et al. [55] |
| deposition | ||||
| 20 | F | ↑ liver and kidney oxidative DNA damage | ||
| 20 | M and F | ↓ expression of detoxification genes in | ||
| liver and spleen | ||||
| 24 | M and F | kidney and liver lipid peroxidation | ||
| 24 | M and F | anti ds-DNA | ||
| C57B6/129SVJ | 48 | F | ↑anti ds-DNA | Li et al. [55] |
| 48 | F | substantial renal glomerular IgG, IgM, C3 | ||
| deposition | ||||
| 48 | F | liver IgG, IgM deposition | ||
| 48 | F | heart, brain IgG, IgM, C3 deposition | ||
| 48 | F | ↑ kidney lipid peroxidation | ||
| 48 | M and F | ↑ liver lipid peroxidation | ||
| 48 | F | ↑ liver and kidney oxidative DNA damage | ||
| 48 | F | ↑ kidney, liver, and spleen cell apoptosis | ||
| 36–48 | F | ↑ spontaneous apoptotic rate in | ||
| splenocytes | ||||
| 129SVJ | 24 | F | ↑ kidney and liver lipid peroxidation | Ma et al. [46] |
| 36 | not | average age for development of | ||
| specified | glomerular lesions | |||
| 48 | F | ↑ kidney and liver lipid peroxidation | ||
| 15; 25; 50; 70 | survival rate (%): 71; 50; 40–45; 20–25 | F | ||
| 15; 25; 50; 70 | M | survival rate (%): 95; 85; 70; 55 |
, not changed or, not different.| Model of SLE | ||||
|---|---|---|---|---|
| Duration | Effect on | |||
| (Mouse Strain) | Tissue/Cells | Nrf2/Nrf2 Targets | Other Pathways | Refs. |
| Pristane | ||||
| 1–2 weeks | peritoneal | ↓Nrf2, Gpx4, Prdx1 | ↓HSP70 protein | Han et al. [48] |
| (C57BL/6) | exudate | Gclc, Nqo1, Sod2, | ↑mitochondrial superoxide | |
| macrophages | Gsr, Srxn gene | |||
| expression | ||||
| ↓Nrf2 binding to | ||||
| ARE-motif | ||||
| 5 months | kidney | HO-1 protein | ↑MCP1 protein | Ebihara et al. [66] |
| (Balb/c) | Nqo1 mRNA | |||
| 5.5 months | Kidney | ↑Nrf2, NQO1 protein | ↑iNos, Tgfb1, Nqo1, Fn mRNA | Jiang et al. [16] |
| 7 months | kidney | ↑Nrf2, NQO1 protein | ↑iNos, Fn mRNA; ↑p-NFκB-p65 | |
| (C57B/SV129) | ||||
| 6 months | Kidney | ↓Nrf2, HO-1 protein | ↑NFκB-p65, p-STAT3 | Castejon et al. [63] |
| (Balb/c) | ↑p-p38, p-JNK, p-ERK | |||
| ↑iNOS; ↑mPGES-1, PGE2 | ||||
| ↑NLRP3, IL-1β, IL-18 | ||||
| 6 months | Kidney | ↓Nrf2, HO-1 protein | ↓IκBα; ↑p-STAT3 | Aparicio- |
| (Balb/c) | ↑mPGES-1, PGE2 | Soto et al. [65] | ||
| ↑p-p38, p-JNK, p-ERK | ||||
| 7 months | Kidney | ↓nuclear Nrf2 | ↑p-NFκB; | Li et al. [64] |
| (Balb/c) | ↓HO-1 protein | ↑NLRP3, cleaved casp1, IL-1β | ||
| ↑ROS, GPx activity | ||||
| 7 months | MDSC | ↓nuclear Nrf2 | ↑Il1b, Il6, Il8, Tnfa mRNA | Li et al. [67] |
| (Balb/c) | (spleen) | HO-1 protein | ↑senescence markers (p21, | |
| p53, p21) | ||||
| ↑iNOS, p47phox | ||||
| ASLN | ||||
| 5 weeks | kidney | ↓nuclear Nrf2 | ↑IL-6, p47phox; ↑GPx activity | Tsai et al. [68] |
| (NZB/NZW) | ||||
| 5 weeks | kidney | ↓nuclear Nrf2 | ↑NLRP3, IL-1β | Ka et al. [69] |
| (NZB/NZW) | ↑p47phox, COX-2, PGE2 | |||
| NZB/W F1 | ||||
| 8.5 months | kidney | ↓nuclear Nrf2 | ↑p47phox; ↑GPx activity | Tsai et al. [70] |
| ↓Ho1, Nqo1 mRNA | ↑NLRP3, IL-1β, cleaved casp1 | |||
| BPA exposure | ||||
| 6 weeks | kidney | ↓Nrf2 protein | ↑NFκB-p65; ↓mTOR | Dong et al. [71] |
| (MRL/lpr) | Abnormal autophagy signaling | |||
| ↑ERα and AhR expression | ||||
| TCE-induced | ||||
| 6 months | liver | Nrf2 protein | NFκB, iNOS | Banerjee et al. [72] |
| p-p38, p-JNK, p-ERK | ||||
| 9 months | ↓Nrf2 protein | ↑NFκB, iNOS | ||
| ↑p-p38, p-JNK, p-ERK | ||||
| ↑IL-12 protein and mRNA | ||||
| ↑protein carbonyls | ||||
| 13 months | ↓Nrf2 protein | ↑NFκB, iNOS | ||
| (MRL/MpJ) | ↑p-p38, p-JNK, p-ERK | |||
| ↑IL-12, TNFα, RANTES protein | ||||
| ↑Il12, Rantes mRNA | ||||
| ↑protein carbonyls | ||||
, not changed or, not different.| Nrf2 Inducer | Model of SLE | Effects | Ref |
|---|---|---|---|
| Sulforaphane | TCE-induced | ↓ p38 and ERK MAPK phosphorylation | Banerjee et al. [71] |
| ↓ Tnfa and Il12 mRNA | |||
| Pristane-induced | ↓ albuminuria | Jiang et al. [16] | |
| Augmented renal Nrf2 and NQO1 protein abundance | |||
| Dimethyl | Pristane-induced | ↓ glomerular injury and proteinuria | Ebihara et al. [66] |
| Fumarate | ↑ HO-1 protein, Nqo1 mRNA | ||
| ↓ MCP1 protein and mRNA; Tgfb and Fn mRNA | |||
| CDDO-Im | Pristane-induced | ↓ classic macrophages in B6 mice | Han et al. [48] |
| ↓ mitochondrial superoxide in macrophages | |||
| ↓ macrophage Ifnar1 and IFN-stimulated gene expression | |||
| Baicalein | Pristane-induced | ↓ anti-dsDNA antibodies, proteinuria, renal injury | Li et al. [64] |
| ↓ serum IL-1B and IL-18, and renal oxidative stress | |||
| ↑ Renal Nrf2 and HO-1 and phospho-NFκB and NLRP3 | |||
| ↓MDSCs in kidney, spleen, bone marrow, and PBMCs | |||
| Extra virgin olive oil | Pristane-induced | restored serum MMP3, renal Nrf2 and HO-1 abundance | Aparicio-Soto |
| attenuated renal p38, ERK, and JNK phosphorylation | et al. [65] | ||
| ↓ LPS-induced TNFα, IL-6, IL-10, and IL-17 in splenocytes | |||
| Oleuropein | Pristane-induced | ↓ inflammatory markers and renal injury, Nrf2 | Castejon et al. [63] |
| Dihydro- | Pristane-induced | inhibit MDSC senescence | Li et al. [67] |
| artemisinin | |||
| BXSB mice | ↓ serum and macrophage secretion of TNFα; renal NFκB | Li et al. [92] | |
| Artemisinin | chronic graft | ↓ proteinuria; ↓ inflammatory, pro-fibrotic mediators | Wu et al. [93] |
| vs. host disease | |||
| Artesunate | MRL/lpr | ↓ anti-dsDNA; ↓ proteinuria; improved kidney function | Jin et al. [94] |
| ↓ renal Il6, Ifn, and Il21 mRNA | Dang et al. [95] | ||
| SM934 | MRL/lpr | ↓ anti-dsDNA; ↓ renal injury, proteinuria, serum BUN | Hou et al. [96] |
| ↓ IL-6, Il-10, Il-12, activated B cells and plasma cells | Wu et al. [97] | ||
| Antroquinonol | ASLN mice | ↓ proteinuria, hematuria, kidney injury | Tsai et al. [68] |
| improve kidney function | |||
| ↑ Nrf2 activity and ↓ ROS in kidney | |||
| Citral | ASLN mice | ↓ proteinuria, renal injury; improved kidney function | Ka et al. [69] |
| ↑ Nrf2 activity; ↓ ROS and NLRP3 inflammasome | |||
| EGCG | NZB/W F1 mice | ↓proteinuria, serum BUN and creatinine, and nephritis | Tsai et al. [70] |
| unaltered glomerular IgG deposition or anti-dsDNA | |||
| restored Nrf2 protein, Nqo1 and Ho1 mRNA | |||
| ↓ inflammasome markers |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barati, M.T.; Caster, D.J. The Potential of Nrf2 Activation as a Therapeutic Target in Systemic Lupus Erythematosus. Metabolites 2022, 12, 151. https://doi.org/10.3390/metabo12020151
Barati MT, Caster DJ. The Potential of Nrf2 Activation as a Therapeutic Target in Systemic Lupus Erythematosus. Metabolites. 2022; 12(2):151. https://doi.org/10.3390/metabo12020151
Chicago/Turabian StyleBarati, Michelle T., and Dawn J. Caster. 2022. "The Potential of Nrf2 Activation as a Therapeutic Target in Systemic Lupus Erythematosus" Metabolites 12, no. 2: 151. https://doi.org/10.3390/metabo12020151
APA StyleBarati, M. T., & Caster, D. J. (2022). The Potential of Nrf2 Activation as a Therapeutic Target in Systemic Lupus Erythematosus. Metabolites, 12(2), 151. https://doi.org/10.3390/metabo12020151