
MetAMDB: Metabolic Atom Mapping Database


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
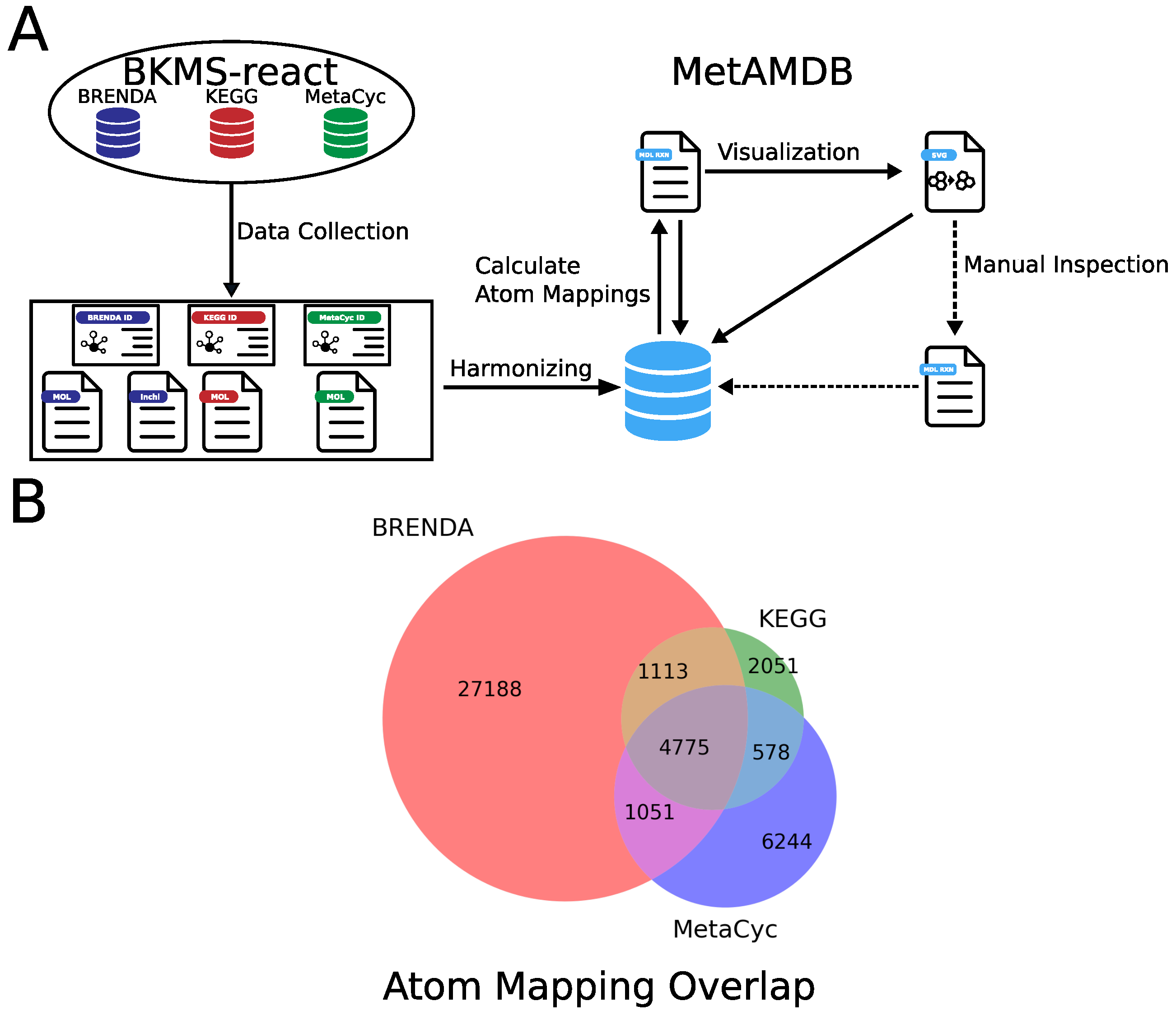
2. Data Collection and Processing
2.1. Collection of Reaction and Metabolite Data
2.2. Automated Generation of Atom Mappings
2.3. Automatic Generation of Atom Mapping Models
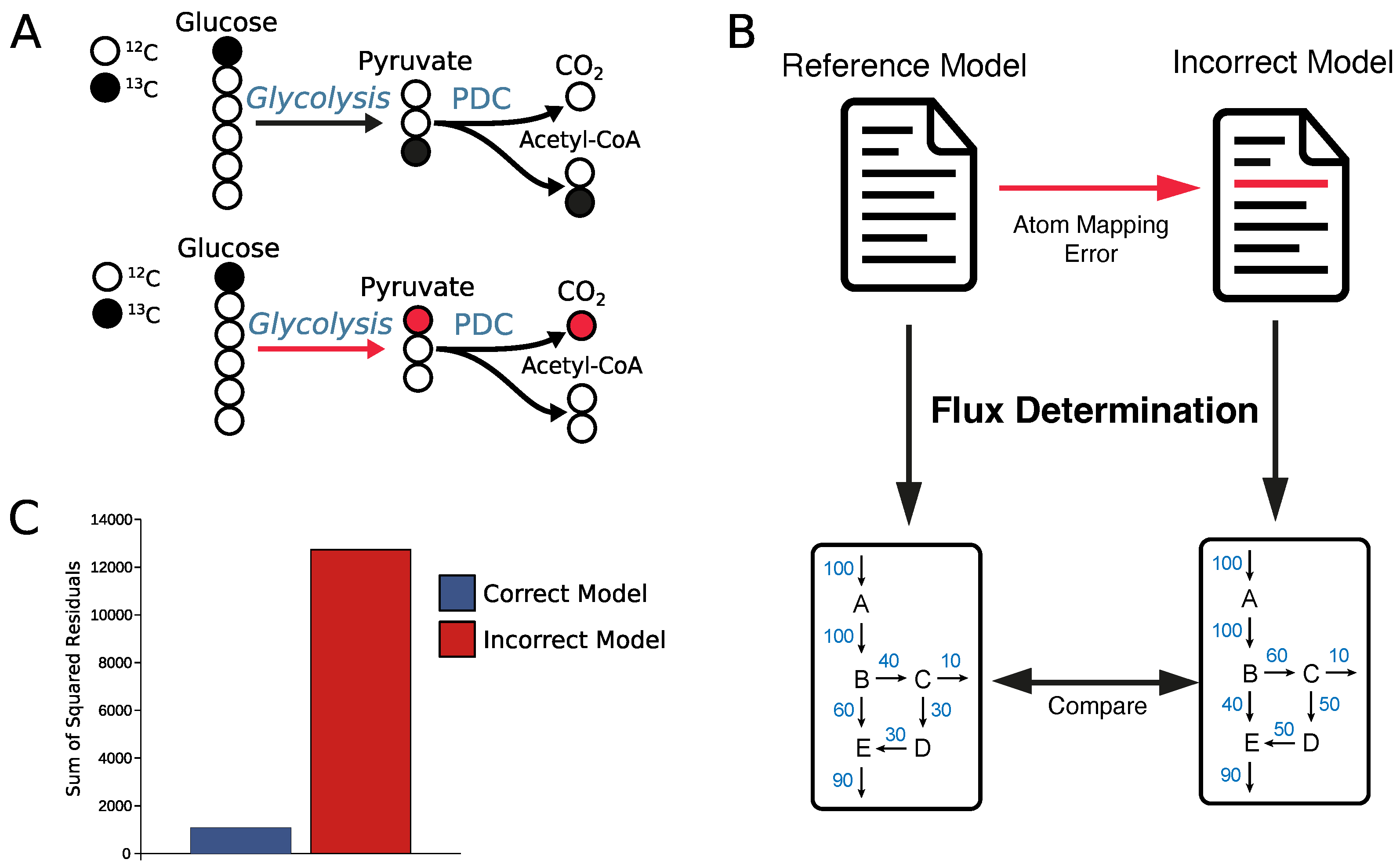
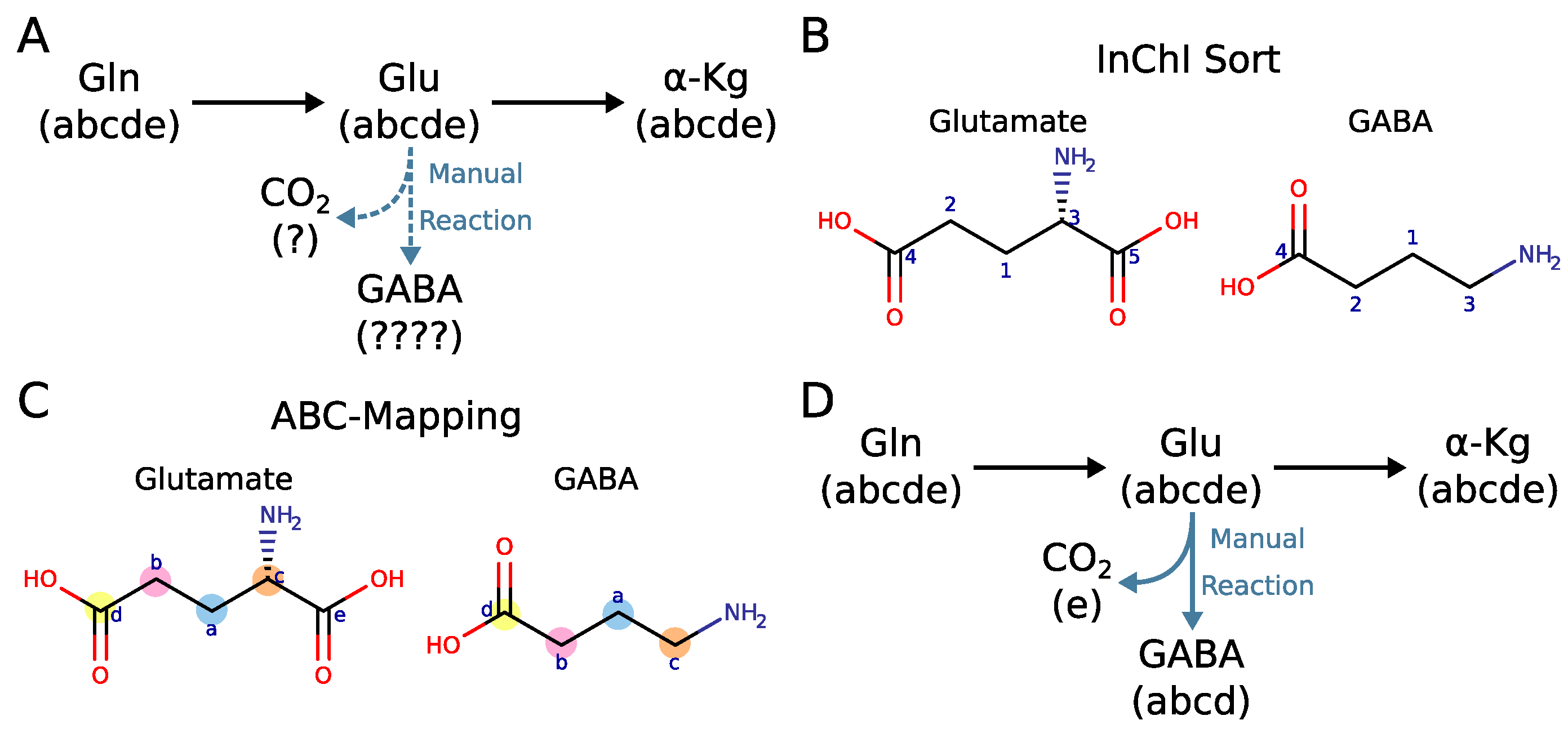
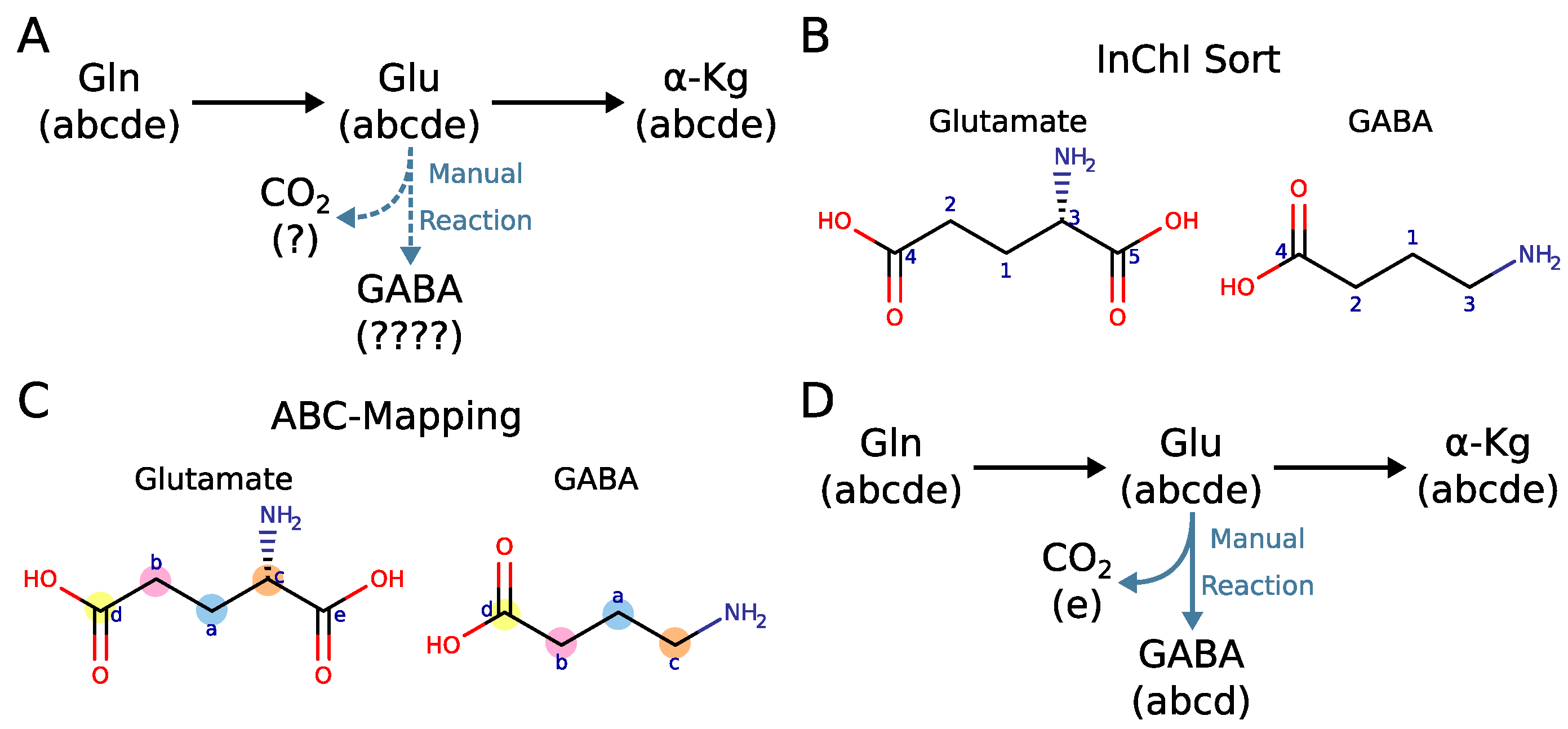
2.4. Why Curation of Atom Mappings Is Needed
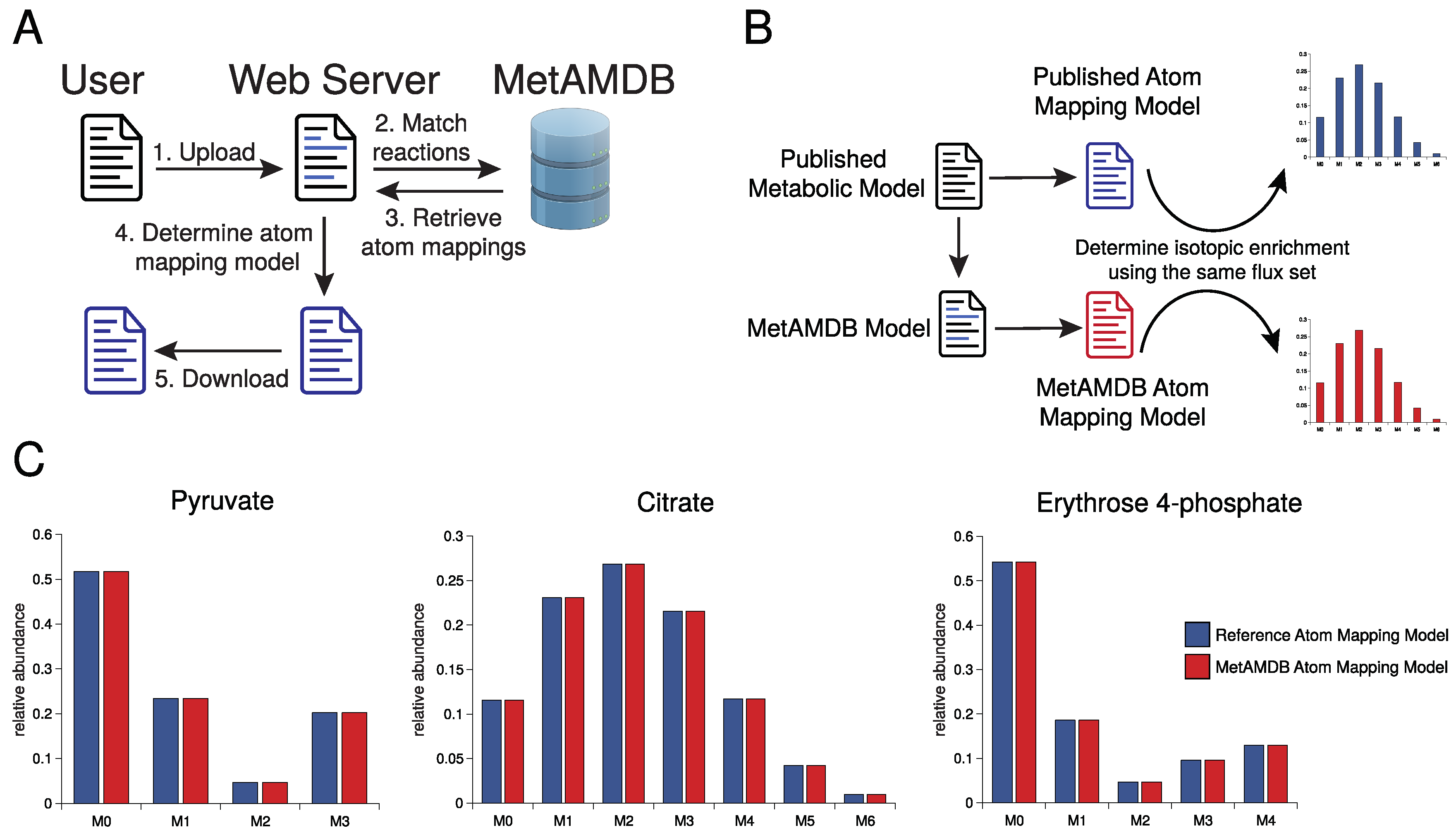
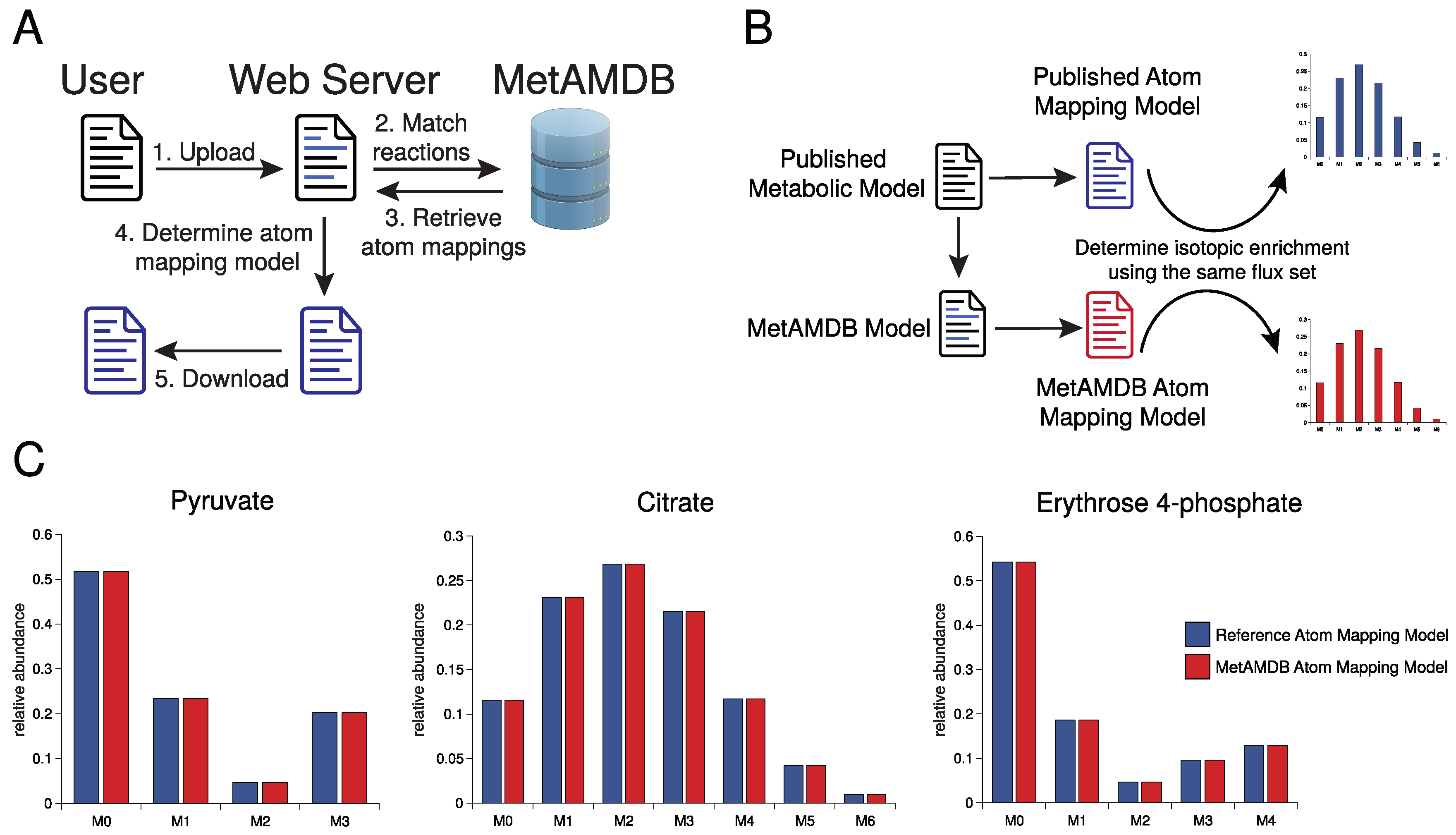
3. Database Content and Usage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wegner, A.; Meiser, J.; Weindl, D.; Hiller, K. How metabolites modulate metabolic flux. Curr. Opin. Biotechnol. 2015, 34, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniewicz, M. A guide to metabolic flux analysis in metabolic engineering: Methods, tools and applications. Metab. Eng. 2021, 63, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.∅. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, G. Metabolic fluxes and metabolic engineering. Metab. Eng. 1999, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lakrisenko, P.; Weindl, D. Dynamic models for metabolomics data integration. Curr. Opin. Syst. Biol. 2021, 28, 100358. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med. 2018, 50, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preciat Gonzalez, G.; El Assal, L.; Noronha, A.; Thiele, I.; Haraldsdóttir, H.; Fleming, R. Comparative evaluation of atom mapping algorithms for balanced metabolic reactions: Application to Recon 3D. J. Cheminform. 2017, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Gonzalez, G.A.P.; Aurich, M.K.; et al. Recon3D: A Resource Enabling A Three-Dimensional View of Gene Variation in Human Metabolism. Nat. Biotechnol. 2018, 36, 272. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.; Kothari, A.; Krummenacker, M.; Midford, P.; Ong, W.; Paley, S.; Subhraveti, P.; Karp, P. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2019, 47, D542–D549. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Altman, T.; Travers, M.; Kothari, A.; Caspi, R.; Karp, P.D. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinform. 2013, 14, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Moseley, H.N. Hierarchical Harmonization of Atom-Resolved Metabolic Reactions across Metabolic Databases. Metabolites 2021, 11, 431. [Google Scholar] [CrossRef]
- First, E.; Gounaris, C.; Floudas, C. Stereochemically consistent reaction mapping and identification of multiple reaction mechanisms through integer linear optimization. J. Chem. Inf. Model. 2012, 52, 84–92. [Google Scholar] [CrossRef]
- Kumar, A.; Maranas, C. CLCA: Maximum common molecular substructure queries within the MetRxn database. J. Chem. Inf. Model. 2014, 54, 3417–3438. [Google Scholar] [CrossRef]
- Latendresse, M.; Malerich, J.; Travers, M.; Karp, P. Accurate atom-mapping computation for biochemical reactions. J. Chem. Inf. Model. 2012, 52, 2970–2982. [Google Scholar] [CrossRef]
- Rahman, S.; Torrance, G.; Baldacci, L.; Martínez Cuesta, S.; Fenninger, F.; Gopal, N.; Choudhary, S.; May, J.; Holliday, G.; Steinbeck, C.; et al. Reaction Decoder Tool (RDT): Extracting features from chemical reactions. Bioinformatics 2016, 32, 2065–2066. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, W.; Szymkuć, S.; Mikulak-Klucznik, B.; Piecuch, K.; Klucznik, T.; Kaźmierowski, M.; Rydzewski, J.; Gambin, A.; Grzybowski, B.A. Automatic mapping of atoms across both simple and complex chemical reactions. Nat. Commun. 2019, 10, 1434. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Kotera, M.; Okuno, Y.; Hattori, M.; Goto, S.; Kanehisa, M. Computational assignment of the EC numbers for genomic-scale analysis of enzymatic reactions. J. Am. Chem. Soc. 2004, 126, 16487–16498. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Walther, J.L.; Antoniewicz, M.R.; Yoo, H.; Stephanopoulos, G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 2008, 99, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Young, J. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starke, C.; Wegner, A. MetAMDB: Metabolic Atom Mapping Database. Metabolites 2022, 12, 122. https://doi.org/10.3390/metabo12020122
Starke C, Wegner A. MetAMDB: Metabolic Atom Mapping Database. Metabolites. 2022; 12(2):122. https://doi.org/10.3390/metabo12020122
Chicago/Turabian StyleStarke, Collin, and Andre Wegner. 2022. "MetAMDB: Metabolic Atom Mapping Database" Metabolites 12, no. 2: 122. https://doi.org/10.3390/metabo12020122
APA StyleStarke, C., & Wegner, A. (2022). MetAMDB: Metabolic Atom Mapping Database. Metabolites, 12(2), 122. https://doi.org/10.3390/metabo12020122