


Alteration of Autophagy and Glial Activity in Nilotinib-Treated Huntington’s Disease Patients

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Nilotinib Is Safe and Tolerated in HD Patients
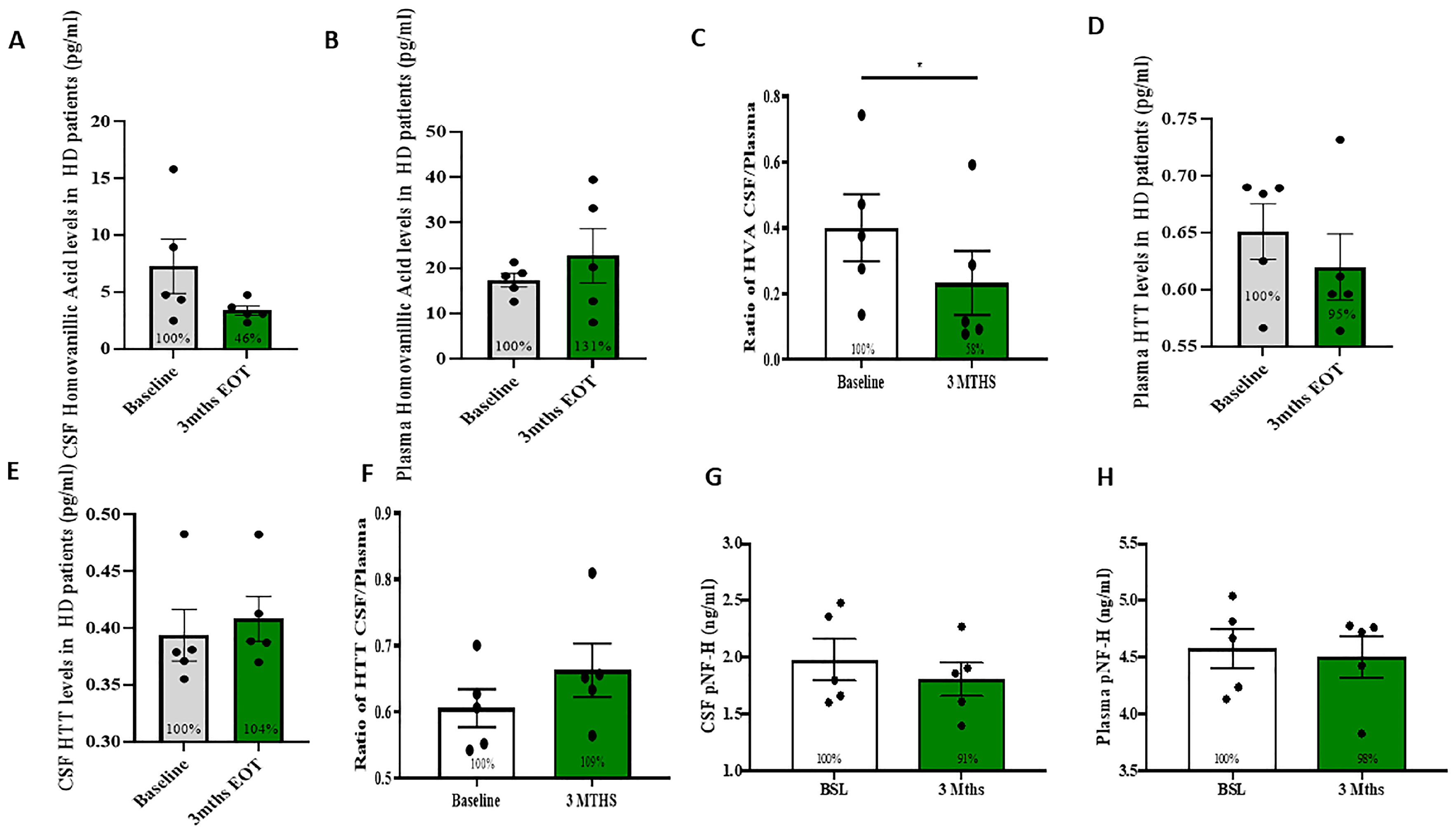
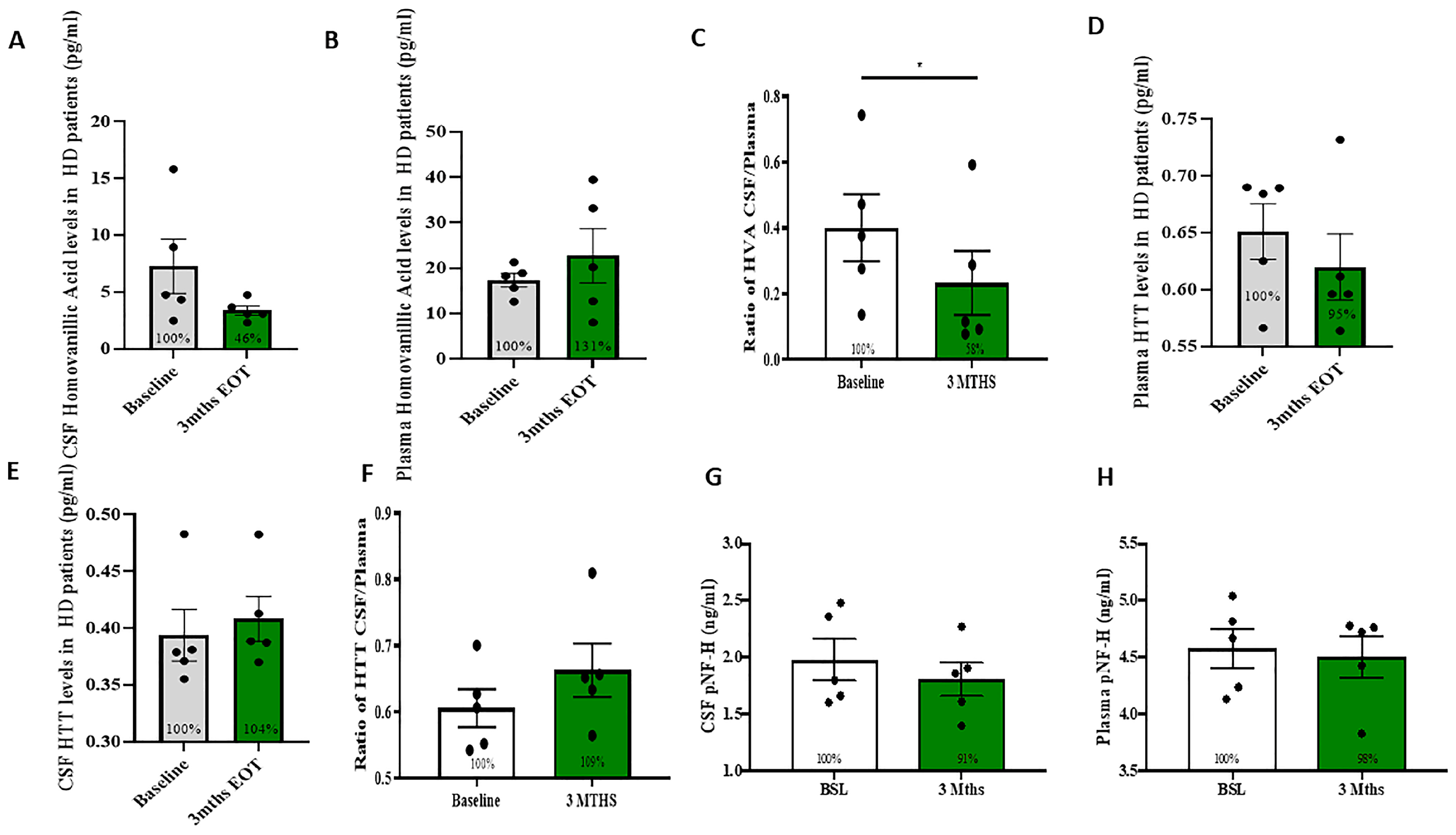
2.2. Measurement of DA Metabolites and HTT
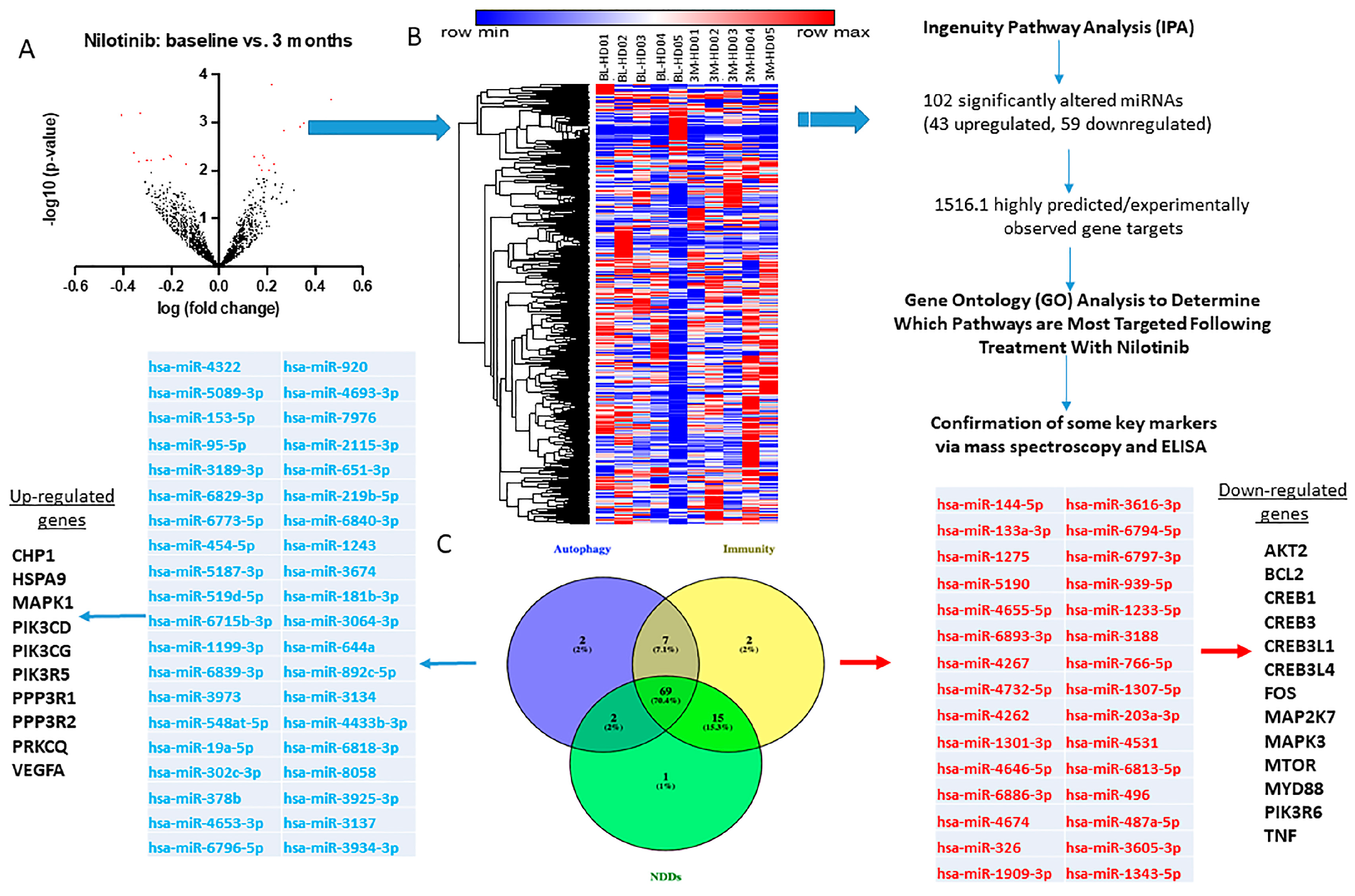
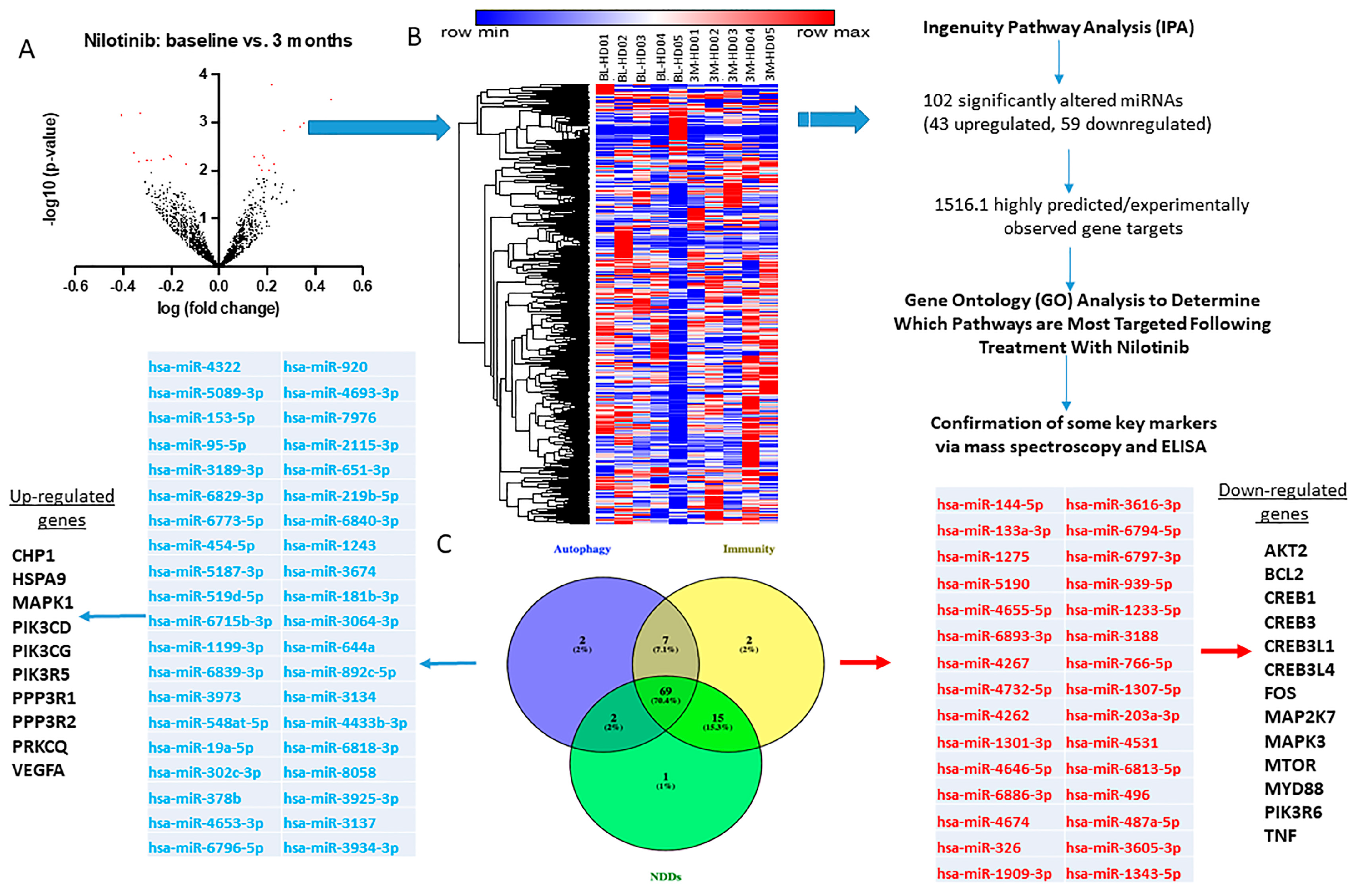
2.3. CSF miRNAs Expression Are Differentially Altered at Baseline and 3 Months in HD
2.4. miRNAs That Control Neurotransmitters of Basal Ganglia Pathways Are Significantly Altered
2.5. Longitudinal Alterations of miRNAs That Regulate Autophagy in HD
2.6. Alterations of miRNAs Targeting Genes That Regulate Sirtuin Pathways and Microglial Activity
2.7. Alterations of miRNAs Targeting Genes Associated with Neurodegenerative Disease Processes
2.8. Exploratory Clinical Effects
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Study Approval
4.3. Cerebrospinal Fluid (CSF) Collection and HVA Measurement
4.4. Neuro-Inflammatory Markers Panel
4.5. microRNA Sequencing
4.6. microRNA Expression Analysis and Gene Ontology
4.7. Posthoc Power Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Vonsattel, J.P.; Richardson, E.P., Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1988, 47, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Graveland, G.A.; Williams, R.S.; DiFiglia, M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 1985, 227, 770–773. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Kowall, N.W.; Richardson, E.P., Jr. Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: A combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J. Neurosci. 1991, 11, 3877–3887. [Google Scholar] [CrossRef]
- Vale, T.C.; Cardoso, F. Chorea: A Journey through History. Tremor Other Hyperkinetic Mov. 2015, 5, tre-5-296. [Google Scholar] [CrossRef]
- Eddy, C.M.; Parkinson, E.G.; Rickards, H.E. Changes in mental state and behaviour in Huntington’s disease. Lancet Psychiatry 2016, 3, 1079–1086. [Google Scholar] [CrossRef]
- Anderson, K.E. Huntington’s disease. Handb. Clin. Neurol. 2011, 100, 15–24. [Google Scholar] [CrossRef]
- Ross, C.A.; Pantelyat, A.; Kogan, J.; Brandt, J. Determinants of functional disability in Huntington’s disease: Role of cognitive and motor dysfunction. Mov. Disord. 2014, 29, 1351–1358. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.K.; Eddy, C. The role of dopamine and glutamate modulation in Huntington disease. Behav Neurol 2013, 26, 255–263. [Google Scholar] [CrossRef]
- Yu-Taeger, L.; Stricker-Shaver, J.; Arnold, K.; Bambynek-Dziuk, P.; Novati, A.; Singer, E.; Lourhmati, A.; Fabian, C.; Magg, J.; Riess, O.; et al. Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells 2019, 8, 595. [Google Scholar] [CrossRef] [Green Version]
- Crook, Z.R.; Housman, D.E. Dysregulation of dopamine receptor D2 as a sensitive measure for Huntington disease pathology in model mice. Proc. Natl. Acad. Sci. USA 2012, 109, 7487–7492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J., Jr.; Sibley, D.R. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Song, W.J.; Yan, Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J. Neurosci. 1996, 16, 6579–6591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, J.L.; Wegrzynowicz, M.; Aschner, M.; Bowman, A.B. Disease-toxicant interactions in manganese exposed Huntington disease mice: Early changes in striatal neuron morphology and dopamine metabolism. PLoS ONE 2012, 7, e31024. [Google Scholar] [CrossRef] [Green Version]
- Deremer, D.L.; Ustun, C.; Natarajan, K. Nilotinib: A second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin. Ther. 2008, 30, 1956–1975. [Google Scholar] [CrossRef]
- Fowler, A.J.; Hebron, M.; Missner, A.A.; Wang, R.; Gao, X.; Kurd-Misto, B.T.; Liu, X.; Moussa, C.E. Multikinase Abl/DDR/Src Inhibition Produces Optimal Effects for Tyrosine Kinase Inhibition in Neurodegeneration. Drugs RD 2019, 19, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Jeitany, M.; Leroy, C.; Tosti, P.; Lafitte, M.; Le Guet, J.; Simon, V.; Bonenfant, D.; Robert, B.; Grillet, F.; Mollevi, C.; et al. Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Mol. Med. 2018, 10, e7918. [Google Scholar] [CrossRef]
- Day, E.; Waters, B.; Spiegel, K.; Alnadaf, T.; Manley, P.W.; Buchdunger, E.; Walker, C.; Jarai, G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. [Google Scholar] [CrossRef]
- Manley, P.W.; Drueckes, P.; Fendrich, G.; Furet, P.; Liebetanz, J.; Martiny-Baron, G.; Mestan, J.; Trappe, J.; Wartmann, M.; Fabbro, D. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim. Biophys. Acta 2010, 1804, 445–453. [Google Scholar] [CrossRef]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Parkinsons Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J.; et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2019, 77, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Arellano, J.; Howard, H.H.; et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharmacol. Res. Perspect. 2019, 7, e00470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, W.; Brakebusch, C.; Fassler, R.; Alves, F.; Ruggiero, F.; Pawson, T. Discoidin domain receptor 1 is activated independently of beta(1) integrin. J. Biol. Chem. 2000, 275, 5779–5784. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Xing, D.; Lu, Z.; Fan, Y.; Hou, W.; Dong, H.; Xiong, L.; Dong, H. DDR1 may play a key role in destruction of the blood-brain barrier after cerebral ischemia-reperfusion. Neurosci. Res. 2015, 96, 14–19. [Google Scholar] [CrossRef]
- Fowler, A.J.; Hebron, M.; Balaraman, K.; Shi, W.; Missner, A.A.; Greenzaid, J.D.; Chiu, T.L.; Ullman, C.; Weatherdon, E.; Duka, V.; et al. Discoidin Domain Receptor 1 is a therapeutic target for neurodegenerative diseases. Hum. Mol. Genet. 2020, 29, 2882–2898. [Google Scholar] [CrossRef]
- Hebron, M.; Peyton, M.; Liu, X.; Gao, X.; Wang, R.; Lonskaya, I.; Moussa, C.E. Discoidin domain receptor inhibition reduces neuropathology and attenuates inflammation in neurodegeneration models. J. Neuroimmunol. 2017, 311, 1–9. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E. Tyrosine kinase inhibition facilitates autophagic SNCA/alpha-synuclein clearance. Autophagy 2013, 9, 1249–1250. [Google Scholar] [CrossRef] [Green Version]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [Green Version]
- Lonskaya, I.; Desforges, N.M.; Hebron, M.L.; Moussa, C.E. Ubiquitination increases parkin activity to promote autophagic alpha-synuclein clearance. PLoS ONE 2013, 8, e83914. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Javidnia, M.; Moussa, C.E. Tau clearance improves astrocytic function and brain glutamate-glutamine cycle. J. Neurol. Sci. 2018, 391, 90–99. [Google Scholar] [CrossRef]
- Pagan, F.L.; Wilmarth, B.; Torres-Yaghi, Y.; Hebron, M.L.; Mulki, S.; Ferrante, D.; Matar, S.; Ahn, J.; Moussa, C. Long-Term Safety and Clinical Effects of Nilotinib in Parkinson’s Disease. Mov. Disord. 2020, 36, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef]
- Saftig, P.; Beertsen, W.; Eskelinen, E.L. LAMP-2: A control step for phagosome and autophagosome maturation. Autophagy 2008, 4, 510–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, A.J.; Moussa, C.E. Activating Autophagy as a Therapeutic Strategy for Parkinson’s Disease. CNS Drugs 2018, 32, 1–11. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Algarzae, N.K.; Desforges, N.; Moussa, C.E. Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson’s disease. Neuroscience 2013, 232, 90–105. [Google Scholar] [CrossRef] [Green Version]
- Vandenbark, A.A.; Offner, H.; Matejuk, S.; Matejuk, A. Microglia and astrocyte involvement in neurodegeneration and brain cancer. J. Neuroinflamm. 2021, 18, 298. [Google Scholar] [CrossRef]
- Turner, R.S.; Hebron, M.L.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, J.N.; Anjum, M.; Pagan, F.; Torres-Yaghi, Y.; Shi, W.; et al. Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer’s Disease. Ann. Neurol. 2020, 88, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Barajas, C.; Coronel, I.; Floran, B. Dopamine Receptors and Neurodegeneration. Aging Dis. 2015, 6, 349–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forn, J.; Krueger, B.K.; Greengard, P. Adenosine 3’,5’-monophosphate content in rat caudate nucleus: Demonstration of dopaminergic and adrenergic receptors. Science 1974, 186, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Greengard, P.; Allen, P.B.; Nairn, A.C. Beyond the dopamine receptor: The DARPP-32/protein phosphatase-1 cascade. Neuron 1999, 23, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 2015, 19, 2540–2548. [Google Scholar] [CrossRef]
- Geva, M.; Kusko, R.; Soares, H.; Fowler, K.D.; Birnberg, T.; Barash, S.; Wagner, A.M.; Fine, T.; Lysaght, A.; Weiner, B.; et al. Pridopidine activates neuroprotective pathways impaired in Huntington Disease. Hum. Mol. Genet. 2016, 25, 3975–3987. [Google Scholar] [CrossRef] [Green Version]
- Grachev, I.D.; Meyer, P.M.; Becker, G.A.; Bronzel, M.; Marsteller, D.; Pastino, G.; Voges, O.; Rabinovich, L.; Knebel, H.; Zientek, F.; et al. Sigma-1 and dopamine D2/D3 receptor occupancy of pridopidine in healthy volunteers and patients with Huntington disease: A [(18)F] fluspidine and [(18)F] fallypride PET study. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1103–1115. [Google Scholar] [CrossRef]
- Gaura, V.; Lavisse, S.; Payoux, P.; Goldman, S.; Verny, C.; Krystkowiak, P.; Damier, P.; Supiot, F.; Bachoud-Levi, A.C.; Remy, P. Association Between Motor Symptoms and Brain Metabolism in Early Huntington Disease. JAMA Neurol. 2017, 74, 1088–1096. [Google Scholar] [CrossRef] [Green Version]
- Mochel, F.; Durant, B.; Durr, A.; Schiffmann, R. Altered dopamine and serotonin metabolism in motorically asymptomatic R6/2 mice. PLoS ONE 2011, 6, e18336. [Google Scholar] [CrossRef] [Green Version]
- Humbert, S.; Bryson, E.A.; Cordelieres, F.P.; Connors, N.C.; Datta, S.R.; Finkbeiner, S.; Greenberg, M.E.; Saudou, F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2002, 2, 831–837. [Google Scholar] [CrossRef]
- Rangone, H.; Pardo, R.; Colin, E.; Girault, J.A.; Saudou, F.; Humbert, S. Phosphorylation of arfaptin 2 at Ser260 by Akt Inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J. Biol. Chem. 2005, 280, 22021–22028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colin, E.; Regulier, E.; Perrin, V.; Durr, A.; Brice, A.; Aebischer, P.; Deglon, N.; Humbert, S.; Saudou, F. Akt is altered in an animal model of Huntington’s disease and in patients. Eur. J. Neurosci. 2005, 21, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Ahn, J.; Hebron, M.; Chiu, T.; Ayoub, R.; Mulki, S.; Ressom, H.; Torres-Yaghi, Y.; Wilmarth, B.; Pagan, F.L.; et al. CSF MicroRNAs Reveal Impairment of Angiogenesis and Autophagy in Parkinson Disease. Neurol. Genet. 2021, 7, e633. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Shekoyan, A.R.; Hebron, M.L.; Desforges, N.; Algarzae, N.K.; Moussa, C.E. Diminished parkin solubility and co-localization with intraneuronal amyloid-beta are associated with autophagic defects in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 231–247. [Google Scholar] [CrossRef]
- Rub, U.; Vonsattel, J.P.; Heinsen, H.; Korf, H.W. The Neuropathology of Huntington s disease: Classical findings, recent developments and correlation to functional neuroanatomy. Adv. Anat. Embryol. Cell Biol. 2015, 217, 1–146. [Google Scholar]
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef] [Green Version]
- Manjula, R.; Anuja, K.; Alcain, F.J. SIRT1 and SIRT2 Activity Control in Neurodegenerative Diseases. Front. Pharmacol. 2020, 11, 585821. [Google Scholar] [CrossRef]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Syed, A.; Lukacsovich, T.; Purcell, J.; Barbaro, B.A.; Worthge, S.A.; Wei, S.R.; Pollio, G.; Magnoni, L.; Scali, C.; et al. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum. Mol. Genet. 2014, 23, 2995–3007. [Google Scholar] [CrossRef] [Green Version]
- Dubinsky, J.M. Towards an Understanding of Energy Impairment in Huntington’s Disease Brain. J. Huntington’s Dis. 2017, 6, 267–302. [Google Scholar] [CrossRef] [Green Version]
- Mochel, F.; Charles, P.; Seguin, F.; Barritault, J.; Coussieu, C.; Perin, L.; Le Bouc, Y.; Gervais, C.; Carcelain, G.; Vassault, A.; et al. Early energy deficit in Huntington disease: Identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007, 2, e647. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.; Hou, G.; Ahmad, P.J.; Fu, E.Y.; Koh, L.; Vogel, W.F.; Bendeck, M.P. Discoidin domain receptor 1 (ddr1) deletion decreases atherosclerosis by accelerating matrix accumulation and reducing inflammation in low-density lipoprotein receptor-deficient mice. Circ. Res. 2008, 102, 1202–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, C.; Magnusson-Lind, A.; Lu, G.; Wagner, P.K.; Southwell, A.L.; Hayden, M.R.; Bjorkqvist, M.; Leavitt, B.R. Enhanced immune response to MMP3 stimulation in microglia expressing mutant huntingtin. Neuroscience 2016, 325, 74–88. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Orr, M.; Liu, P. Sample size estimation while controlling false discovery rate for microarray experiments using ssize.fdr package. R J. 2009, 1, 47–53. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, K.E.; Stevenson, M.; Varghese, R.; Hebron, M.L.; Koppel, E.; McCartin, M.; Kuprewicz, R.; Matar, S.; Ferrante, D.; Moussa, C. Alteration of Autophagy and Glial Activity in Nilotinib-Treated Huntington’s Disease Patients. Metabolites 2022, 12, 1225. https://doi.org/10.3390/metabo12121225
Anderson KE, Stevenson M, Varghese R, Hebron ML, Koppel E, McCartin M, Kuprewicz R, Matar S, Ferrante D, Moussa C. Alteration of Autophagy and Glial Activity in Nilotinib-Treated Huntington’s Disease Patients. Metabolites. 2022; 12(12):1225. https://doi.org/10.3390/metabo12121225
Chicago/Turabian StyleAnderson, Karen E., Max Stevenson, Rency Varghese, Michaeline L. Hebron, Erin Koppel, Mara McCartin, Robin Kuprewicz, Sara Matar, Dalila Ferrante, and Charbel Moussa. 2022. "Alteration of Autophagy and Glial Activity in Nilotinib-Treated Huntington’s Disease Patients" Metabolites 12, no. 12: 1225. https://doi.org/10.3390/metabo12121225
APA StyleAnderson, K. E., Stevenson, M., Varghese, R., Hebron, M. L., Koppel, E., McCartin, M., Kuprewicz, R., Matar, S., Ferrante, D., & Moussa, C. (2022). Alteration of Autophagy and Glial Activity in Nilotinib-Treated Huntington’s Disease Patients. Metabolites, 12(12), 1225. https://doi.org/10.3390/metabo12121225