
Familial Hypercholesterolemia and Lipoprotein(a): A Gordian Knot in Cardiovascular Prevention
 , , ,
, , , 

Abstract
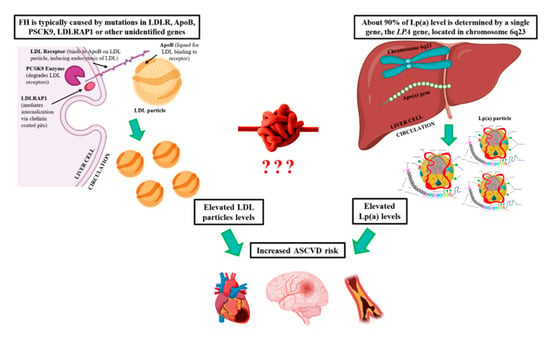
1. Introduction
2. Materials and Methods
3. Results
3.1. Familial Hypercholesterolemia
3.1.1. Definition and Prevalence of FH
3.1.2. Genetics of FH
3.1.3. Clinical Presentation of FH
3.1.4. Diagnosis of FH
3.1.5. Prognosis and Treatment of FH
3.2. Lipoprotein(a)
3.2.1. Molecular Properties of Lp(a)
3.2.2. Genetics of Lp(a)
3.2.3. Definition and Prevalence of High Lp(a)
3.2.4. Clinical Presentation of hyperLp(a)
3.2.5. Available and Upcoming Therapies for hyperLp(a)
4. Discussion
4.1. FH and hyperLp(a)
4.1.1. Prevalence of hyperLp(a) in FH Patients

{kind=link}
| Authors | Sample Size | Country | Diagnosis of FH | Results |
|---|---|---|---|---|
| Utermann et al. [62] | 381 | UK | Clinical | 102 FH patients vs. 279 healthy subjects: 41.3 vs. 14.1 mg/dL, p < 0.001. |
| Langsted et al. [7] | 46,200 | Denmark | Clinical and Genetic | 42,934 Unlike vs. 1675 Possible vs. 184 Probable/Definite FH patients: 23 (22.8–23.3) vs. 32 (31–34) vs. 35 (29–41) mg/dL, p < 0.05 (using unadjusted LDL-C for Lp(a)-cholesterol). 43,699 Unlike vs. 2360 Possible vs. 141 Probable/Definite FH patients: 24 (23.5–24.1) vs. 22 (21–24) vs. 21 (16–26) mg/dL, p = 0.46 (after adjusting LDL-C for Lp(a)-cholesterol). Lp(a) concentrations were similar in those with and without FH mutations: 24 (23.4–14) mg/dL in 46,124 individuals without an FH mutation vs. 23 (9–36) mg/dL in 27 individuals with an LDLR mutation (p = 0.10 vs. no known mutation) vs. 21 (14–28) mg/dL in 49 individuals with an apolipoprotein B mutation (p = 0.52) and 22 (15–28) mg/dL in 76 individuals with any FH mutation (p = 0.64) |
| Chan et al. [54] | 907 | N/A | Clinical and Genetic | 74 patients with FH (8.2%) were reclassified to unlike FH when LDL-C was corrected for Lp(a)-cholesterol. There were no significant differences detected in the proportion of pathogenic FH mutations (27.9% vs. 33.1%) between patients with increased and normal Lp(a) concentrations at a cutoff of 50 mg/dL (p = 0.05). |
| Trinder et al. [8] | 37,877 | UK | Clinical and Genetic | British Columbia FH and Familial Combined Hyperlipidemia cohort; 391 FH patients vs. 245 non-FH patients: 28.7 (10.3–75.4) vs. 13 (10–48.9) mg/dL, p < 0.01. No significant differences were noted between carriers of a pathogenic variant in the LDLR or apolipoprotein B and noncarriers (1.43 log mg/dL vs. 1.42 log mg/dL, p = 0.97). UK Biobank cohort (n = 37,486); 221 patients with FH mutation vs. 37,265 without FH mutation: 10.7 (4.9–26.3) vs. 8.7 (4.0–25.8) mg/dL, p = 0.24. |
| Kraft et al. [60] | 69 | South Africa | Clinical and Genetic | 26 HoFH patients vs. 43 HeFH relatives: 36.6 vs. 14.4 mg/dL, p = 0.004. |
| Li et al. [51] | 8050 | China | Clinical and Genetic | 6250 Unlikely vs. 1519 Possible vs. 281 Probable/Definite FH patients: 51.8 vs. 57.1 vs. 60.5 mg/dL, p < 0.001. |
| Leitersdorf et al. [63] | 216 | N/A | Clinical and Genetic | 99 FH patients vs. 117 controls: 33 vs. 22 mg/dL, p < 0.001. |
| Mbewu et al. [64] | 277 | UK | Clinical and Genetic | 89 HeFH patients vs. 109 normocholesterolemic controls vs. 40 healthy controls: 22.7 vs. 10.0 vs. 9.1 mg/dL, p < 0.05. |
| Alonso et al. [6] | 2917 | Spain | Genetic | 1960 HeFH patients vs. 957 non-FH relatives: 23.6 (9.6–59.2) vs. 21.0 (7–47.2) mg/dL, p < 0.001. 500 FH patients with null mutations vs. 246 FH patients with defect LDLR mutations: 24.4 vs. 21.5 mg/dL, p < 0.05. |
| Tada et al. [56] | 4255 | Japan | Genetic | 198 FH patients with LDLR variants vs. 42 with PCSK9 variants vs. 4015 controls: 12.6 (9.4–33.9) vs. 21.1 (11.7–34.9) vs. 5 (2.7–8.1) mg/dL, p = 0.002 for the comparison between FH-LDLR or FH-PCSK9 with control group. |
| Sun et al. [57] | 510 | China | Genetic | 259 HeFH patients vs. 255 matched non-FH controls: 28.9 (13.2–64.8) vs. 11.7 (5.3–26.9) mg/dL, p < 0.05. |
| Guo et al. [59] | 48 | France | Genetic | 8 HoFH patients vs. 18 healthy subjects: 50 ± 32 vs. 20.6 ± 5.2 mg/dL, p < 0.001. |
| Sjouke et al. [61] | 119 | Netherlands | Genetic | 22 unaffected relatives vs. 63 HeFH vs. 34 HoFH patients: 19.9 (11.1–41.5) vs. 24.4 (5.9–70.6) vs. 47.3 (14.9–111.7) mg/dL, p = 0.150. |
| Lingenhel et al. [65] | 203 | South Africa | Genetic | 103 FH patients vs. 100 non-FH relatives: 35.4 ± 31 vs. 20.7 ± 18.1 mg/dL, p = 0.0014. |
| Wiklund et al. [66] | 120 | Sweden | N/A | 47 HeFH patients vs. 47 controls matched for age and sex: 2.4 (2.5–124.5) vs. 9.7 (0.7–104) mg/dL), p < 0.001. |
4.1.2. Joint Effect of FH and hyperLp(a) on Cardiovascular Risk
4.1.3. Barriers to the Identification of FH
4.1.4. Cascade Lp(a) Testing in FH Patients
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bianconi, V.; Banach, M.; Pirro, M. Why patients with familial hypercholesterolemia are at high cardiovascular risk? Beyond LDL-C levels. Trends Cardiovasc. Med. 2021, 31, 205–215. [Google Scholar] [CrossRef]
- Vallejo-Vaz, A.J.; Stevens, C.A.; Lyons, A.R.; Dharmayat, K.I.; Freiberger, T.; Hovingh, G.K.; Mata, P.; Raal, F.J.; Santos, R.D.; Soran, H.; et al. Global perspective of familial hypercholesterolaemia: A cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet 2021, 398, 1713–1725. [Google Scholar] [CrossRef]
- Gencer, B.; Kronenberg, F.; Stroes, E.S.; Mach, F. Lipoprotein(a): The revenant. Eur. Heart J. 2017, 38, 1553–1560. [Google Scholar] [CrossRef]
- Koutsogianni, A.-D.; Liberopoulos, E.; Tselepis, A.D. Lipoprotein(a): An update on its role in human health and disease. J. Atheroscler. Prev. Treat. 2021, 12, 92–102. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.; Ginsberg, H.; Amarenco, P.; Catapano, A.L.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Alonso, R.; Andres, E.; Mata, N.; Fuentes-Jiménez, F.; Badimón, L.; López-Miranda, J.; Padró, T.; Muñiz, O.; Díaz-Díaz, J.L.; Mauri, M.; et al. Lipoprotein(a) Levels in Familial Hypercholesterolemia: An Important Predictor of Cardiovascular Disease Independent of the Type of LDL Receptor Mutation. J. Am. Coll. Cardiol. 2014, 63, 1982–1989. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: A prospective cohort study. Lancet Diabetes Endocrinol. 2016, 4, 577–587. [Google Scholar] [CrossRef]
- Trinder, M.; DeCastro, M.L.; Azizi, H.; Cermakova, L.; Jackson, L.M.; Frohlich, J.; Mancini, G.J.; Francis, G.A.; Brunham, L.R. Ascertainment Bias in the Association Between Elevated Lipoprotein(a) and Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2020, 75, 2682–2693. [Google Scholar] [CrossRef]
- Sjouke, B.; Kusters, D.M.; Kindt, I.; Besseling, J.; Defesche, J.C.; Sijbrands, E.J.; van Lennep, J.E.R.; Stalenhoef, A.F.; Wiegman, A.; de Graaf, J.; et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: Prevalence, genotype–phenotype relationship, and clinical outcome. Eur. Heart J. 2015, 36, 560–565. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: A HuGE Prevalence Review. Am. J. Epidemiol. 2004, 160, 407–420. [Google Scholar] [CrossRef]
- Futema, M.; Whittall, R.A.; Kiley, A.; Steel, L.K.; Cooper, J.A.; Badmus, E.; Leigh, S.E.; Karpe, F.; Neil, H.A.W.; Humphries, S.E. Analysis of the frequency and spectrum of mutations recognised to cause familial hypercholesterolaemia in routine clinical practice in a UK specialist hospital lipid clinic. Atherosclerosis 2013, 229, 161–168. [Google Scholar] [CrossRef]
- Goldberg, A.C.; Hopkins, P.N.; Toth, P.P.; Ballantyne, C.M.; Rader, D.J.; Robinson, J.G.; Daniels, S.R.; Gidding, S.S.; de Ferranti, S.D.; Ito, M.K.; et al. Familial Hypercholesterolemia: Screening, diagnosis and management of pediatric and adult patients: Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 2011, 5, S1–S8. [Google Scholar] [CrossRef]
- Santos, R.D.; Gidding, S.S.; Hegele, R.A.; Cuchel, M.A.; Barter, P.J.; Watts, G.F.; Baum, S.J.; Catapano, A.L.; Chapman, M.J.; Defesche, J.C.; et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: A consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016, 4, 850–861. [Google Scholar] [CrossRef]
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Familial Hypercholesterolemia and Coronary Heart Disease: A HuGE Association Review. Am. J. Epidemiol. 2004, 160, 421–429. [Google Scholar] [CrossRef]
- Hutter, C.M.; Austin, M.A.; Humphries, S.E. Familial Hypercholesterolemia, Peripheral Arterial Disease, and Stroke: A HuGE Minireview. Am. J. Epidemiol. 2004, 160, 430–435. [Google Scholar] [CrossRef]
- Benn, M.; Watts, G.F.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Mutations causative of familial hypercholesterolaemia: Screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur. Heart J. 2016, 37, 1384–1394. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Merz, C.N.B.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef]
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Rizos, C.V.; Skoumas, I.; Rallidis, L.; Skalidis, E.; Tziomalos, K.; Garoufi, A.; Anagnostis, P.; Sfikas, G.; Kotsis, V.; Doumas, M.; et al. LDL cholesterol target achievement in heterozygous familial hypercholesterolemia patients according to 2019 ESC/EAS lipid guidelines: Implications for newer lipid-lowering treatments. Int. J. Cardiol. 2021, 345, 119–124. [Google Scholar] [CrossRef]
- Banach, M.; Rizzo, M.; Obradovic, M.; Montalto, G.; Rysz, J.; Mikhailidis, D.P.; Isenovic, E.R. PCSK9 inhibition-a novel mechanism to treat lipid disorders? Curr. Pharm. Des. 2013, 19, 3869–3877. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Jawi, M.M.; Frohlich, J.; Chan, S.Y. Lipoprotein(a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein(a) Molecule. J. Lipids 2020, 2020, 3491764. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.A. Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Ellis, K.L.; Boffa, M.B.; Sahebkar, A.; Koschinsky, M.L.; Watts, G.F. The renaissance of lipoprotein(a): Brave new world for preventive cardiology? Prog. Lipid Res. 2017, 68, 57–82. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Li, L.; Zhang, Y.; Mo, Y.; Mai, W.; Zhou, Y. Lipoprotein(a): A Promising Marker for Residual Cardiovascular Risk Assessment. Dis. Markers 2013, 35, 551–559. [Google Scholar] [CrossRef][Green Version]
- Saleheen, D.; Haycock, P.C.; Zhao, W.; Rasheed, A.; Taleb, A.; Imran, A.; Abbas, S.; Majeed, F.; Akhtar, S.; Qamar, N.; et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: A mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017, 5, 524–533. [Google Scholar] [CrossRef]
- Hobbs, H.H.; White, A.L. Lipoprotein(a): Intrigues and Insights: Current Opinion in Lipidology. Curr. Opin. Lipidol. 1999, 10, 225–236. Available online: https://journals.lww.com/co-lipidology/Abstract/1999/06000/Lipoprotein_a___intrigues_and_insights.5.aspx (accessed on 21 December 2021).
- Rader, D.J.; Cain, W.; Ikewaki, K.; Talley, G.; Zech, L.A.; Usher, D.; Brewer, H.B. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J. Clin. Investig. 1994, 93, 2758–2763. [Google Scholar] [CrossRef]
- Arai, K.; Luke, M.M.; Koschinsky, M.L.; Miller, E.R.; Pullinger, C.R.; Witztum, J.L.; Kane, J.P.; Tsimikas, S. The I4399M variant of apolipoprotein(a) is associated with increased oxidized phospholipids on apolipoprotein B-100 particles. Atherosclerosis 2010, 209, 498–503. [Google Scholar] [CrossRef]
- Rao, F.; Schork, A.J.; Maihofer, A.X.; Nievergelt, C.M.; Marcovina, S.M.; Miller, E.R.; Witztum, J.L.; O’Connor, D.T.; Tsimikas, S. Heritability of Biomarkers of Oxidized Lipoproteins: A Twin Pair Study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Wilcken, D.E.L.; Dudman, N.P.B. Early expression of the apolipoprotein (a) gene: Relationships between infants’ and their parents’ serum apolipoprotein (a) levels. Pediatrics 1992, 89, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Rifai, N.; Heiss, G.; Doetsch, K. Lipoprotein(a) at birth, in blacks and whites. Atherosclerosis 1992, 92, 123–129. [Google Scholar] [CrossRef]
- Cegla, J.; Neely, R.G.; France, M.; Ferns, G.; Byrne, C.D.; Halcox, J.; Datta, D.; Capps, N.; Shoulders, C.; Qureshi, N.; et al. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis 2019, 291, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef]
- Gencer, B.; Mach, F. Lipoprotein(a): The perpetual supporting actor. Eur. Heart J. 2018, 39, 2597–2599. [Google Scholar] [CrossRef]
- Malaguarnera, G.; Gagliano, C.; Bucolo, C.; Vacante, M.; Salomone, S.; Malaguarnera, M.; Leonardi, D.G.; Motta, M.; Drago, F.; Avitabile, T. Lipoprotein(a) serum levels in diabetic patients with retinopathy. BioMed Res. Int. 2013, 2013, 943505. [Google Scholar] [CrossRef]
- Arnold, M.; Schweizer, J.; Nakas, C.T.; Schütz, V.; Westphal, L.P.; Inauen, C.; Pokorny, T.; Luft, A.; Leichtle, A.; Arnold, M.; et al. Lipoprotein(a) is associated with large artery atherosclerosis stroke aetiology and stroke recurrence among patients below the age of 60 years: Results from the BIOSIGNAL study. Eur. Heart J. 2021, 42, 2186–2196. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Liberopoulos, E.; Tellis, K.; Tselepis, A.D. Oxidized phospholipids and lipoprotein(a): An update. Eur. J. Clin. Investig. 2021, 52, e13710. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef] [PubMed]
- Corrado, E.; Rizzo, M.; Coppola, G.; Muratori, I.; Carella, M.; Novo, S. Endothelial dysfunction and carotid lesions are strong predictors of clinical events in patients with early stages of atherosclerosis: A 24-month follow-up study. Coron. Artery Dis. 2008, 19, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Umemoto, T. Atorvastatin decreases lipoprotein(a): A meta-analysis of randomized trials. Int. J. Cardiol. 2012, 154, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Fraley, A.E.; Schwartz, G.G.; Olsson, A.G.; Kinlay, S.; Szarek, M.; Rifai, N.; Libby, P.; Ganz, P.; Witztum, J.L.; Tsimikas, S. Relationship of Oxidized Phospholipids and Biomarkers of Oxidized Low-Density Lipoprotein With Cardiovascular Risk Factors, Inflammatory Biomarkers, and Effect of Statin Therapy in Patients With Acute Coronary Syndromes: Results From the MIRACL (Myocardial Ischemia Reduction With Aggressive Cholesterol Lowering) Trial. J. Am. Coll. Cardiol. 2009, 53, 2186–2196. [Google Scholar] [CrossRef]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) Concentrations, Rosuvastatin Therapy, and Residual Vascular Risk: An Analysis from the JUPITER Trial. Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Barter, P.; Demicco, D.A.; Bao, W.; Preston, G.M.; LaRosa, J.C.; Grundy, S.M.; Deedwania, P.; Greten, H.; Wenger, N.K.; et al. Prediction of Cardiovascular Events in Statin-Treated Stable Coronary Patients of the Treating to New Targets Randomized Controlled Trial by Lipid and Non-Lipid Biomarkers. PLoS ONE 2014, 9, e114519. [Google Scholar] [CrossRef]
- Naito, R.; Daida, H.; Masuda, D.; Harada-Shiba, M.; Arai, H.; Bujo, H.; Ishibashi, S.; Koga, N.; Oikawa, S.; Yamashita, S. Relation of Serum Lipoprotein(a) Levels to Lipoprotein and Apolipoprotein Profiles and Atherosclerotic Diseases in Japanese Patients with Heterozygous Familial Hypercholesterolemia: Familial Hypercholesterolemia Expert Forum (FAME) Study. J. Atheroscler. Thromb. 2022, 29, 1188–1200. [Google Scholar] [CrossRef]
- Fogacci, F.; Banach, M.; Mikhailidis, D.P.; Bruckert, E.; Toth, P.P.; Watts, G.F.; Reiner, Ž.; Mancini, J.; Rizzo, M.; Mitchenko, O.; et al. Safety of red yeast rice supplementation: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2019, 143, 1–16. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- Li, S.; Wu, N.-Q.; Zhu, C.-G.; Zhang, Y.; Guo, Y.-L.; Gao, Y.; Li, X.-L.; Qing, P.; Cui, C.-J.; Xu, R.-X.; et al. Significance of lipoprotein(a) levels in familial hypercholesterolemia and coronary artery disease. Atherosclerosis 2017, 260, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Liberopoulos, E. Lipoprotein(a) reduction with proprotein convertase subtilisin/kexin type 9 inhibitors: An unsolved mystery. Eur. J. Prev. Cardiol. 2021, 28, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Serban, M.-C.; Mikhailidis, D.P.; Toth, P.P.; Muntner, P.; Ursoniu, S.; Mosterou, S.; Glasser, S.; Martin, S.S.; Jones, S.R.; et al. Head-to-head comparison of statins versus fibrates in reducing plasma fibrinogen concentrations: A systematic review and meta-analysis. Pharmacol. Res. 2016, 103, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Pang, J.; Hooper, A.J.; Bell, D.A.; Burnett, J.R.; Watts, G.F. Effect of Lipoprotein(a) on the Diagnosis of Familial Hypercholesterolemia: Does It Make a Difference in the Clinic? Clin. Chem. 2019, 65, 1258–1266. [Google Scholar] [CrossRef]
- Loh, W.J.; Chan, D.C.; Mata, P.; Watts, G.F. Familial Hypercholesterolemia and Elevated Lipoprotein(a): Cascade Testing and Other Implications for Contextual Models of Care. Front. Genet. 2022, 13, 905941. [Google Scholar] [CrossRef]
- Tada, H.; Kawashiri, M.-A.; Yoshida, T.; Teramoto, R.; Nohara, A.; Konno, T.; Inazu, A.; Mabuchi, H.; Yamagishi, M.; Hayashi, K. Lipoprotein(a) in Familial Hypercholesterolemia With Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Gain-of-Function Mutations. Circ. J. 2016, 80, 512–518. [Google Scholar] [CrossRef]
- Sun, D.; Li, S.; Zhao, X.; Wu, N.-Q.; Zhu, C.-G.; Guo, Y.-L.; Gao, Y.; Qing, P.; Cui, C.-J.; Liu, G.; et al. Association between lipoprotein (a) and proprotein convertase substilisin/kexin type 9 in patients with heterozygous familial hypercholesterolemia: A case-control study. Metabolism 2018, 79, 33–41. [Google Scholar] [CrossRef]
- Luc, G.; Chapman, M.J.; DE Gennes, J.-L.; Turpin, G. A study of the structural heterogeneity of low-density lipoproteins in two patients homozygous for familial hypercholesterolaemia, one of phenotype E2/2. Eur. J. Clin. Investig. 1986, 16, 329–337. [Google Scholar] [CrossRef]
- Guo, H.-C.; Chapman, J.; Bruckert, E.; Farriaux, J.-P.; De Gennes, J.-L. Lipoprotein Lp(a) in homozygous familial hypercholesterolemia: Density profile, particle heterogeneity and apolipoprotein(a) phenotype. Atherosclerosis 1991, 86, 69–83. [Google Scholar] [CrossRef]
- Kraft, H.G.; Lingenhel, A.; Raal, F.J.; Hohenegger, M.; Utermann, G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 522–528. [Google Scholar] [CrossRef]
- Sjouke, B.; Yahya, R.; Tanck, M.W.; Defesche, J.C.; de Graaf, J.; Wiegman, A.; Kastelein, J.J.; Mulder, M.T.; Hovingh, G.K.; van Lennep, J.E.R. Plasma lipoprotein(a) levels in patients with homozygous autosomal dominant hypercholesterolemia. J. Clin. Lipidol. 2017, 11, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Utermann, G.; Hoppichler, F.; Dieplinger, H.; Seed, M.; Thompson, G.; Boerwinkle, E. Defects in the low density lipoprotein receptor gene affect lipoprotein (a) levels: Multiplicative interaction of two gene loci associated with premature atherosclerosis. Proc. Natl. Acad. Sci. USA 1989, 86, 4171–4174. [Google Scholar] [CrossRef] [PubMed]
- Leitersdorf, E.; Friedlander, Y.; Bard, J.; Fruchart, J.; Eisenberg, S.; Stein, Y. Diverse effect of ethnicity on plasma lipoprotein[a] levels in heterozygote patients with familial hypercholesterolemia. J. Lipid Res. 1991, 32, 1513–1519. [Google Scholar] [CrossRef]
- Mbewu, A.D.; Bhatnagar, D.; Durrington, P.N.; Hunt, L.; Ishola, M.; Arrol, S.; Mackness, M.; Lockley, P.; Miller, J.P. Serum lipoprotein(a) in patients heterozygous for familial hypercholesterolemia, their relatives, and unrelated control populations. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Lingenhel, A.; Kraft, H.G.; Kotze, M.; Peeters, A.V.; Kronenberg, F.; Kruse, R.; Utermann, G. Concentrations of the atherogenic Lp(a) are elevated in familial hypercholesterolaemia: A sib pair and family analysis. Eur. J. Hum. Genet. 1998, 6, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, O.; Olofsson, S.-O.; Fager, G.; Bondjers, G.; Angelin, B.; Erikson, M.; Berglund, L. Apolipoprotein(a) and ischaemic heart disease in familial hypercholesterolaemia. Lancet 1990, 335, 1360–1363. [Google Scholar] [CrossRef]
- de Isla, L.P.; Cerezo, A.S.; Alonso, R.; Mata, P. Lipoprotein(a) and familial hypercholesterolaemia. Lancet Diabetes Endocrinol. 2016, 4, 730. [Google Scholar] [CrossRef][Green Version]
- Anagnostis, P.; Rizos, C.V.; Skoumas, I.; Rallidis, L.; Tziomalos, K.; Skalidis, E.; Kotsis, V.; Doumas, M.; Kolovou, G.; Sfikas, G.; et al. Association between lipoprotein(a) concentrations and atherosclerotic cardiovascular disease risk in patients with familial hypercholesterolemia: An analysis from the HELLAS-FH. Endocrine 2022, 76, 324–330. [Google Scholar] [CrossRef]
- Funabashi, S.; Kataoka, Y.; Hori, M.; Ogura, M.; Doi, T.; Noguchi, T.; Harada-Shiba, M. Characterization of Polyvascular Disease in Heterozygous Familial Hypercholesterolemia: Its Association With Circulating Lipoprotein(a) Levels. J. Am. Heart Assoc. 2022, 11, e025232. [Google Scholar] [CrossRef]
- Gudnason, V. Lipoprotein(a): A causal independent risk factor for coronary heart disease? Curr. Opin. Cardiol. 2009, 24, 490–495. [Google Scholar] [CrossRef]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [PubMed]
- Gencer, B.; Dias, K.; Siekmeier, R.; Stojakovic, T. Lipoprotein (a) and Risk of Cardiovascular Disease—A Systematic Review and Meta Analysis of Prospective Studies. Clin. Lab. 2011, 57, 143–156. Available online: https://www.researchgate.net/publication/51058528_Lipoprotein_a_and_Risk_of_Cardiovascular_Disease_-_A_Systematic_Review_and_Meta_Analysis_of_Prospective_Studies (accessed on 22 December 2021).
- Danesh, J.; Collins, R.; Peto, R. Lipoprotein(a) and coronary heart disease. Meta-analysis of prospective studies. Circulation 2000, 102, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Hamasaki, M.; Kotani, K. Risk of cardiovascular disease with lipoprotein(a) in familial hypercholesterolemia: A review. Arch. Med. Sci.-Atheroscler. Dis. 2020, 5, e148. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Bernard, S.; Baass, A. SLC22A3 is associated with lipoprotein (a) concentration and cardiovascular disease in familial hypercholesterolemia. Clin. Biochem. 2019, 66, 44–48. [Google Scholar] [CrossRef]
- Jones, L.K.; Sturm, A.C.; Seaton, T.L.; Gregor, C.; Gidding, S.S.; Williams, M.S.; Rahm, A.K. Barriers, facilitators, and solutions to familial hypercholesterolemia treatment. PLoS ONE 2020, 15, e0244193. [Google Scholar] [CrossRef]
- Chakraborty, A.; Pang, J.; Chan, D.C.; Ellis, K.L.; Hooper, A.J.; Bell, D.A.; Burnett, J.R.; Moses, E.K.; Watts, G.F. Cascade testing for elevated lipoprotein(a) in relatives of probands with familial hypercholesterolaemia and elevated lipoprotein(a). Atherosclerosis 2022, 349, 219–226. [Google Scholar] [CrossRef]
- Ellis, K.L.; Pérez de Isla, L.; Alonso, R.; Fuentes, F.; Watts, G.F.; Mata, P. Value of Measuring Lipoprotein(a) During Cascade Testing for Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 73, 1029–1039. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koutsogianni, A.D.; Adamidis, P.S.; Barkas, F.; Liberopoulos, E.; Su, T.-C.; Yamashita, S.; Liamis, G.; Rizzo, M. Familial Hypercholesterolemia and Lipoprotein(a): A Gordian Knot in Cardiovascular Prevention. Metabolites 2022, 12, 1065. https://doi.org/10.3390/metabo12111065
Koutsogianni AD, Adamidis PS, Barkas F, Liberopoulos E, Su T-C, Yamashita S, Liamis G, Rizzo M. Familial Hypercholesterolemia and Lipoprotein(a): A Gordian Knot in Cardiovascular Prevention. Metabolites. 2022; 12(11):1065. https://doi.org/10.3390/metabo12111065
Chicago/Turabian StyleKoutsogianni, Amalia Despoina, Petros Spyridonas Adamidis, Fotios Barkas, Evangelos Liberopoulos, Ta-Chen Su, Shizuya Yamashita, George Liamis, and Manfredi Rizzo. 2022. "Familial Hypercholesterolemia and Lipoprotein(a): A Gordian Knot in Cardiovascular Prevention" Metabolites 12, no. 11: 1065. https://doi.org/10.3390/metabo12111065
APA StyleKoutsogianni, A. D., Adamidis, P. S., Barkas, F., Liberopoulos, E., Su, T.-C., Yamashita, S., Liamis, G., & Rizzo, M. (2022). Familial Hypercholesterolemia and Lipoprotein(a): A Gordian Knot in Cardiovascular Prevention. Metabolites, 12(11), 1065. https://doi.org/10.3390/metabo12111065