Integrative Analysis of the Metabolome and Transcriptome Provides Insights into the Mechanisms of Flavonoid Biosynthesis in Quinoa Seeds at Different Developmental Stages

Abstract
1. Introduction
2. Results
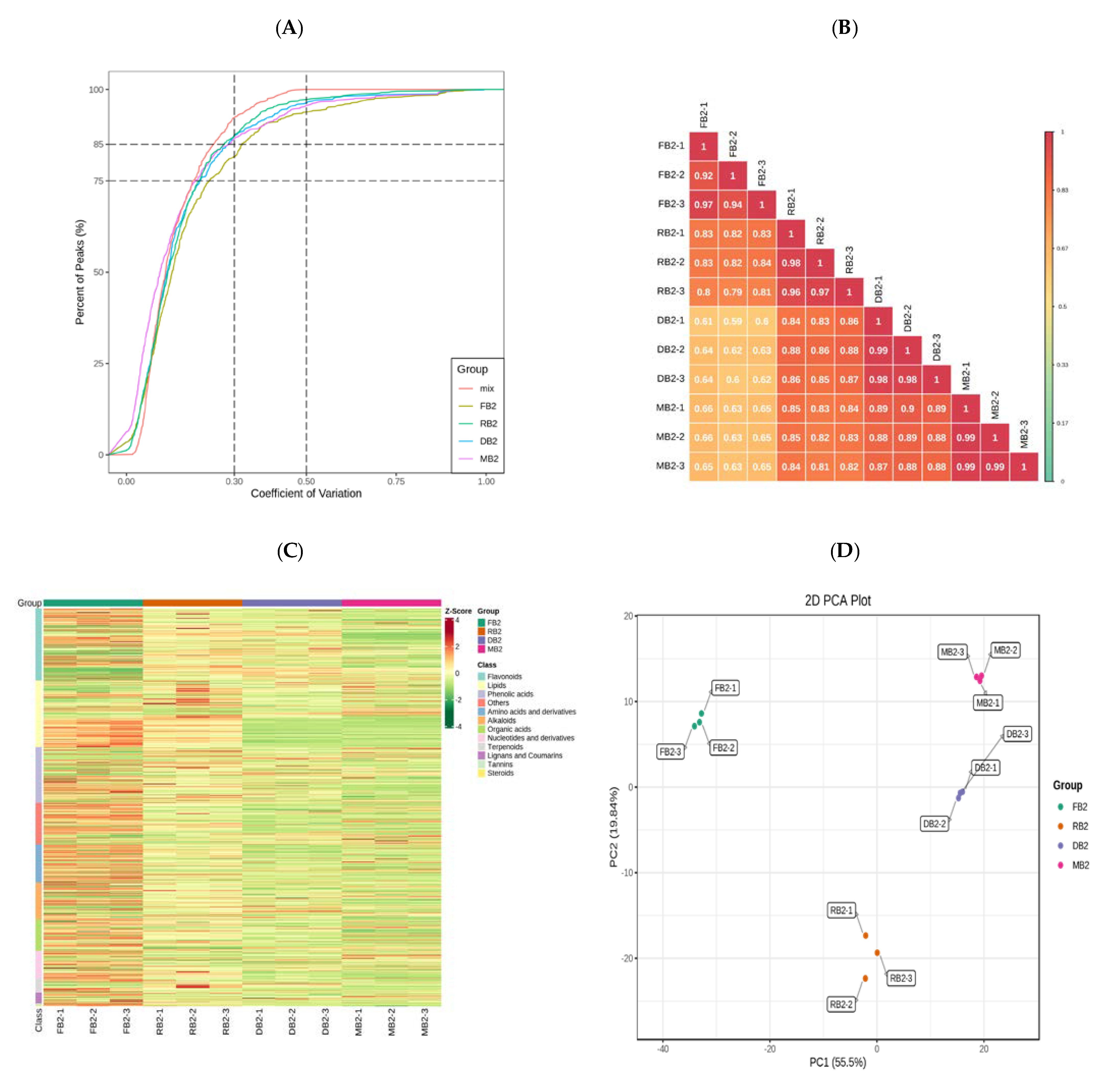
2.1. Qualitative and Quantitative Analyses of Related Metabolites in Quinoa Seeds at Different Development Stages
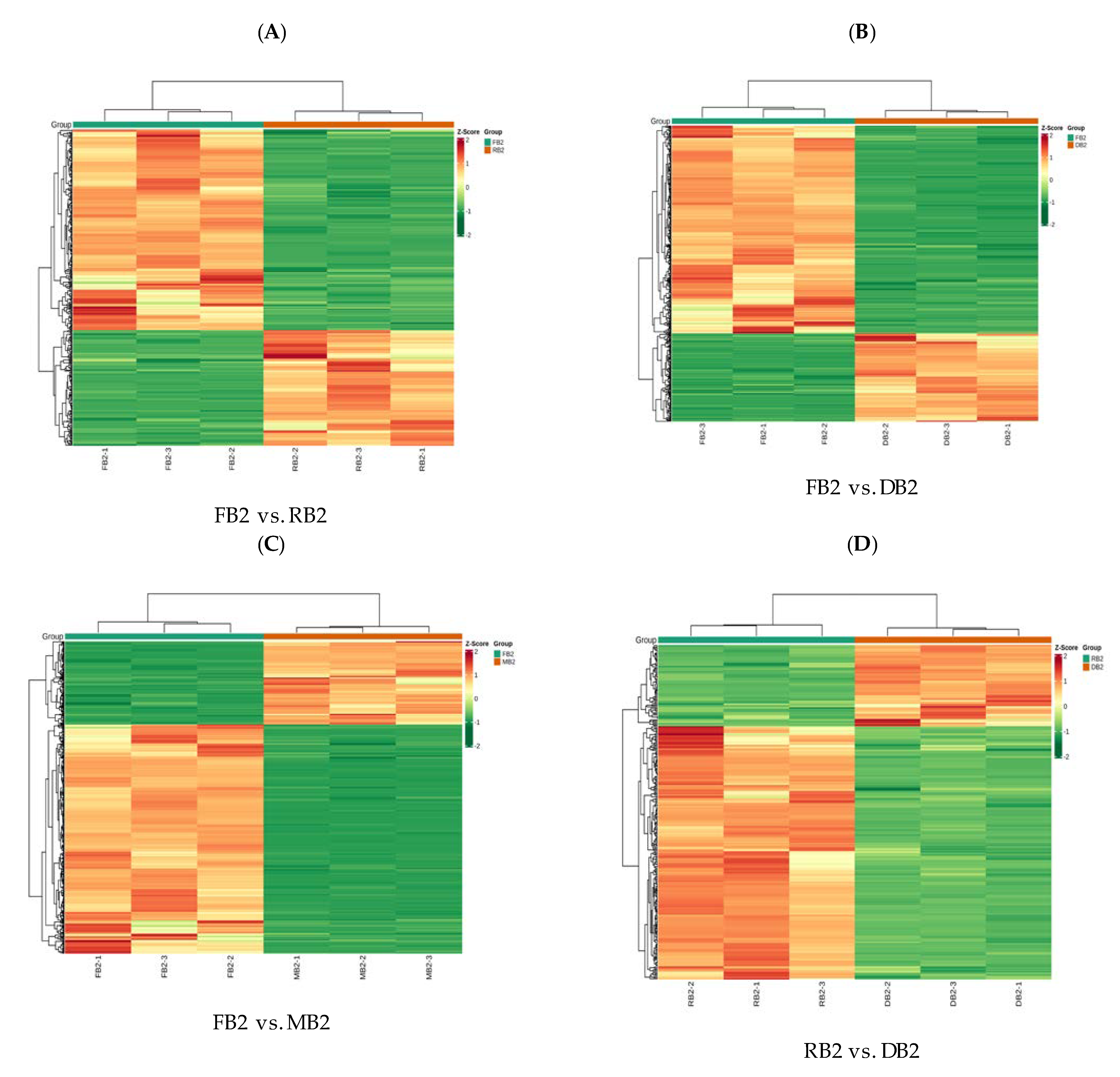
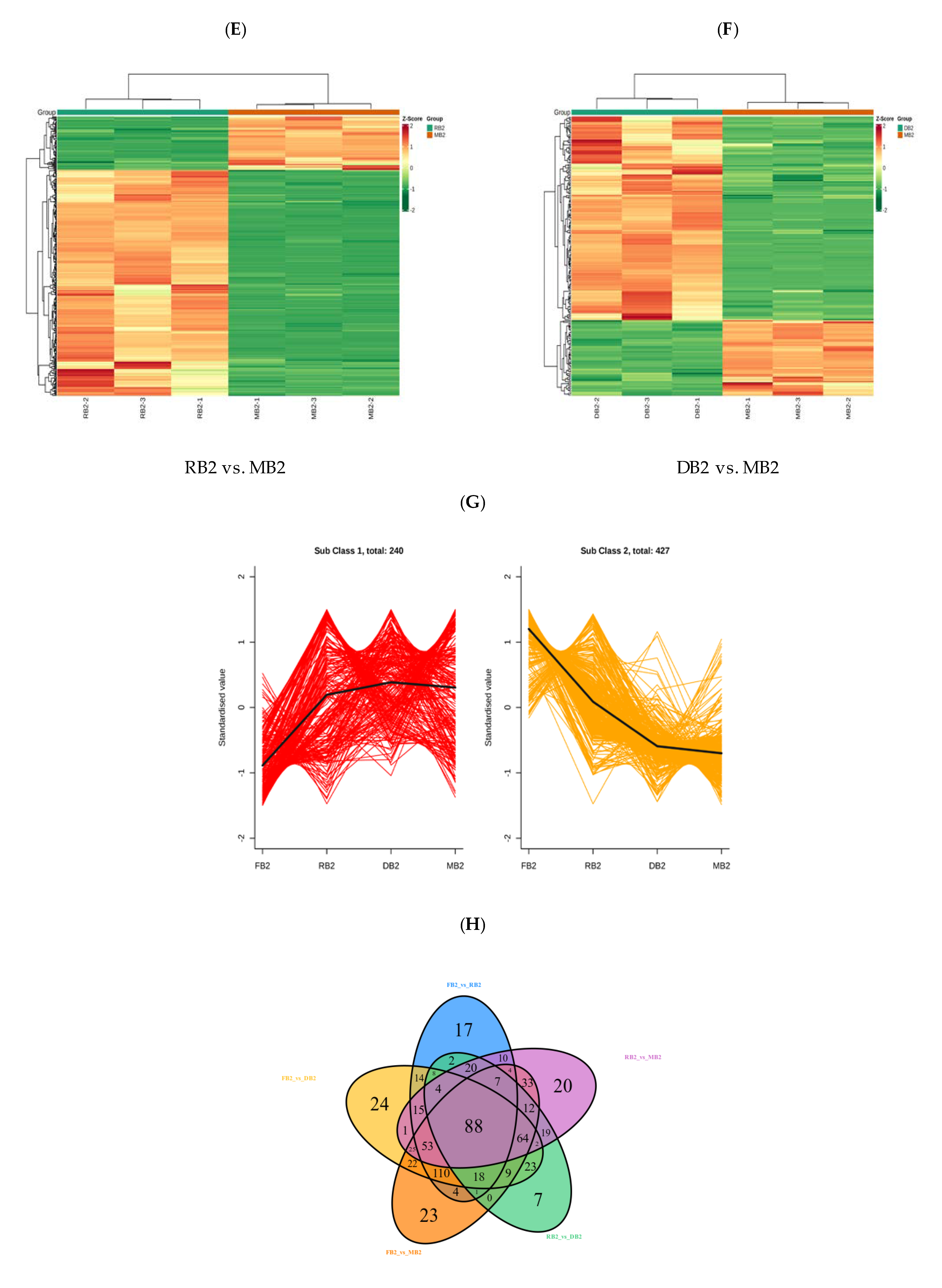
2.2. Analysis of Differences in Flavonoid-Related Metabolites in Quinoa Seeds at Different Developmental Stages
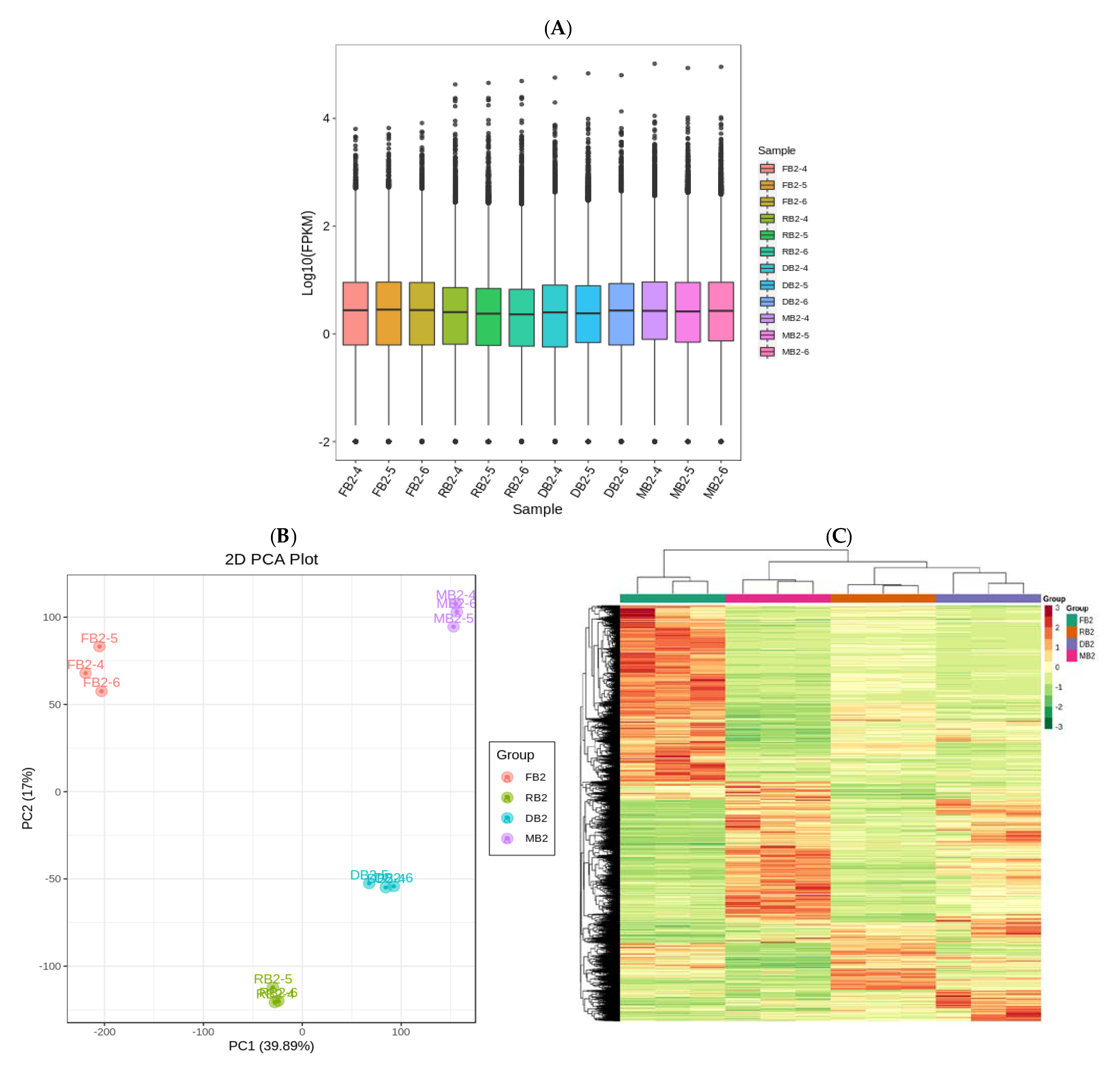
2.3. Transcriptome Analysis of Quinoa Seeds at Different Developmental Stages
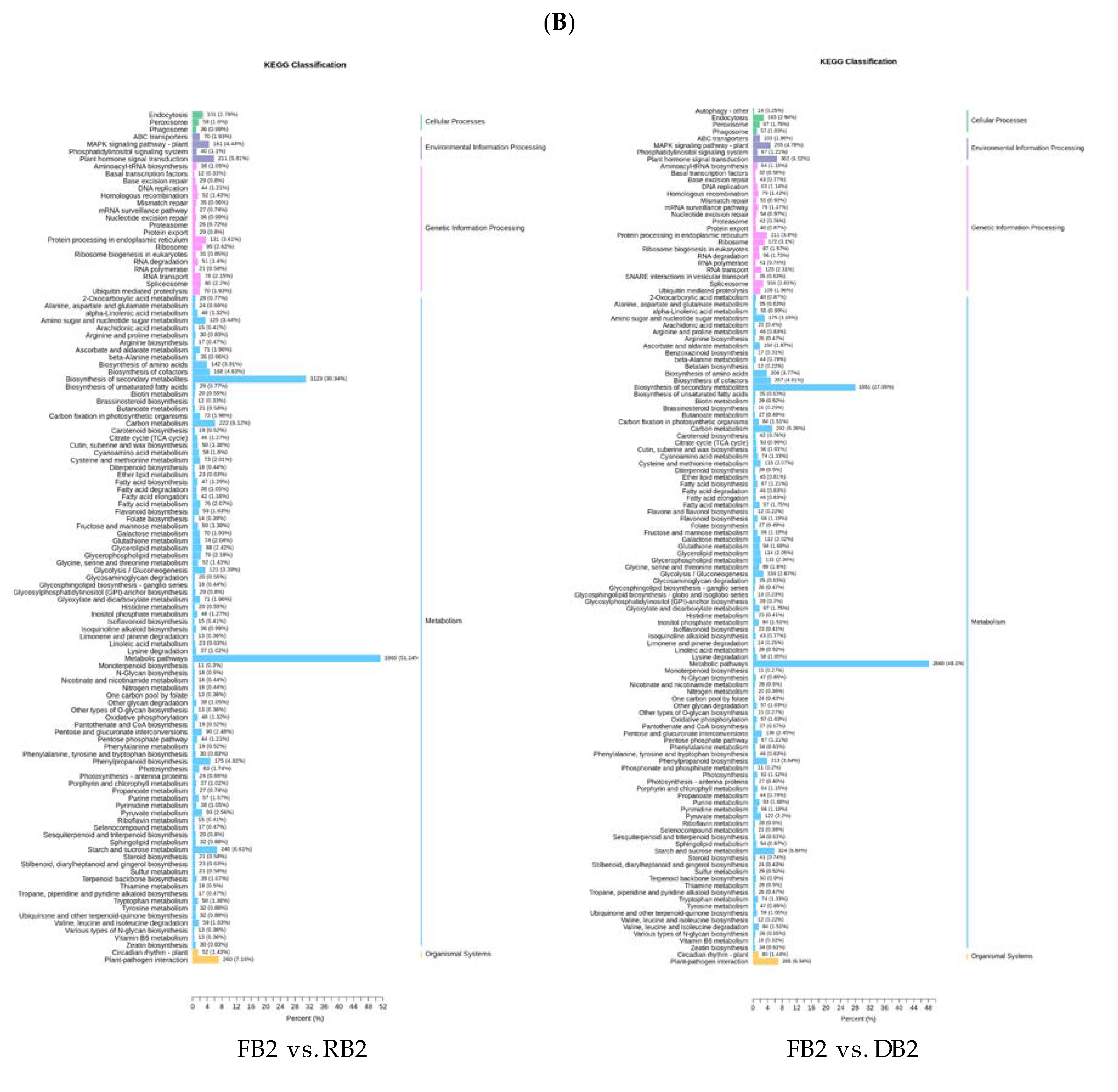
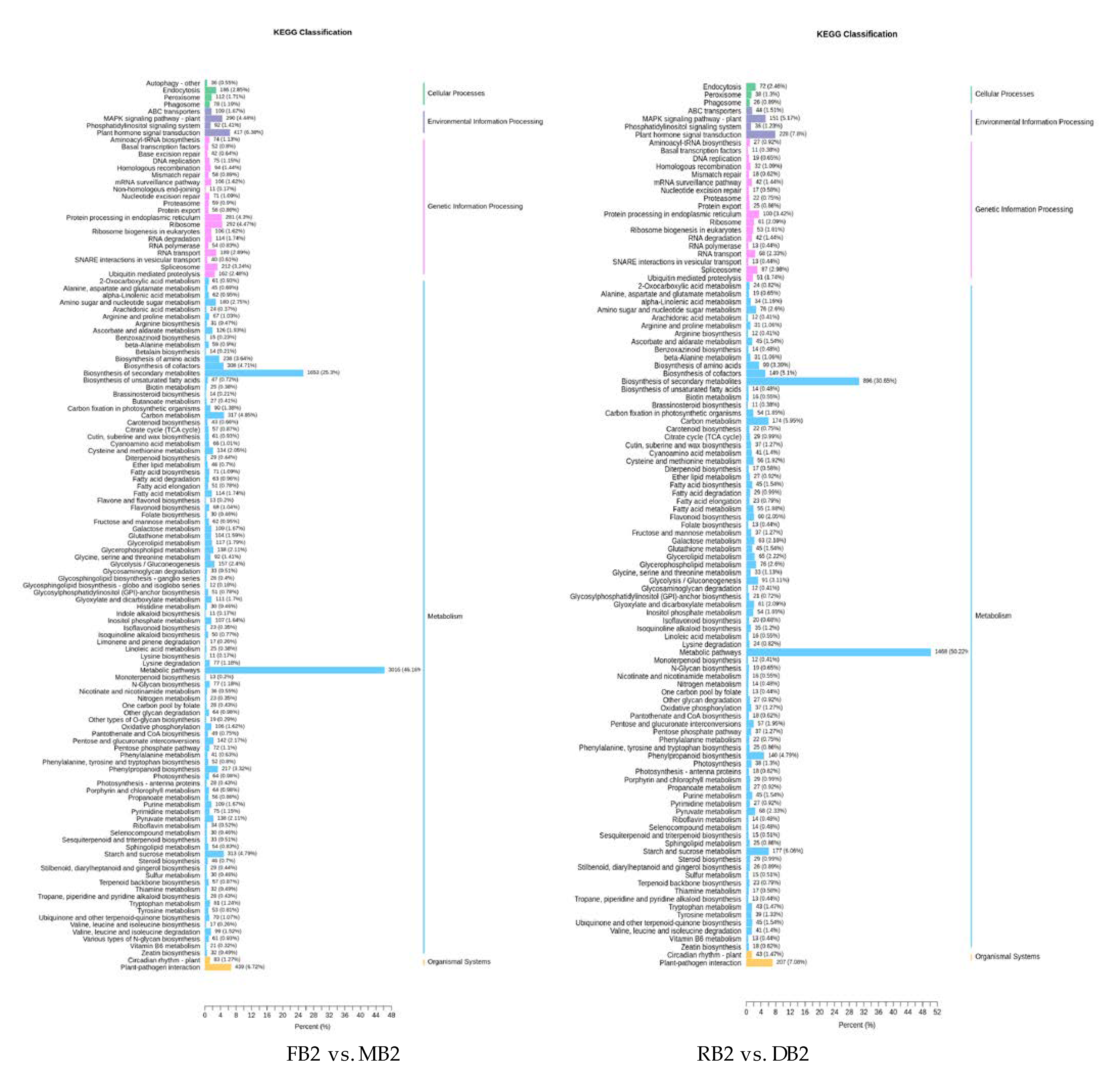
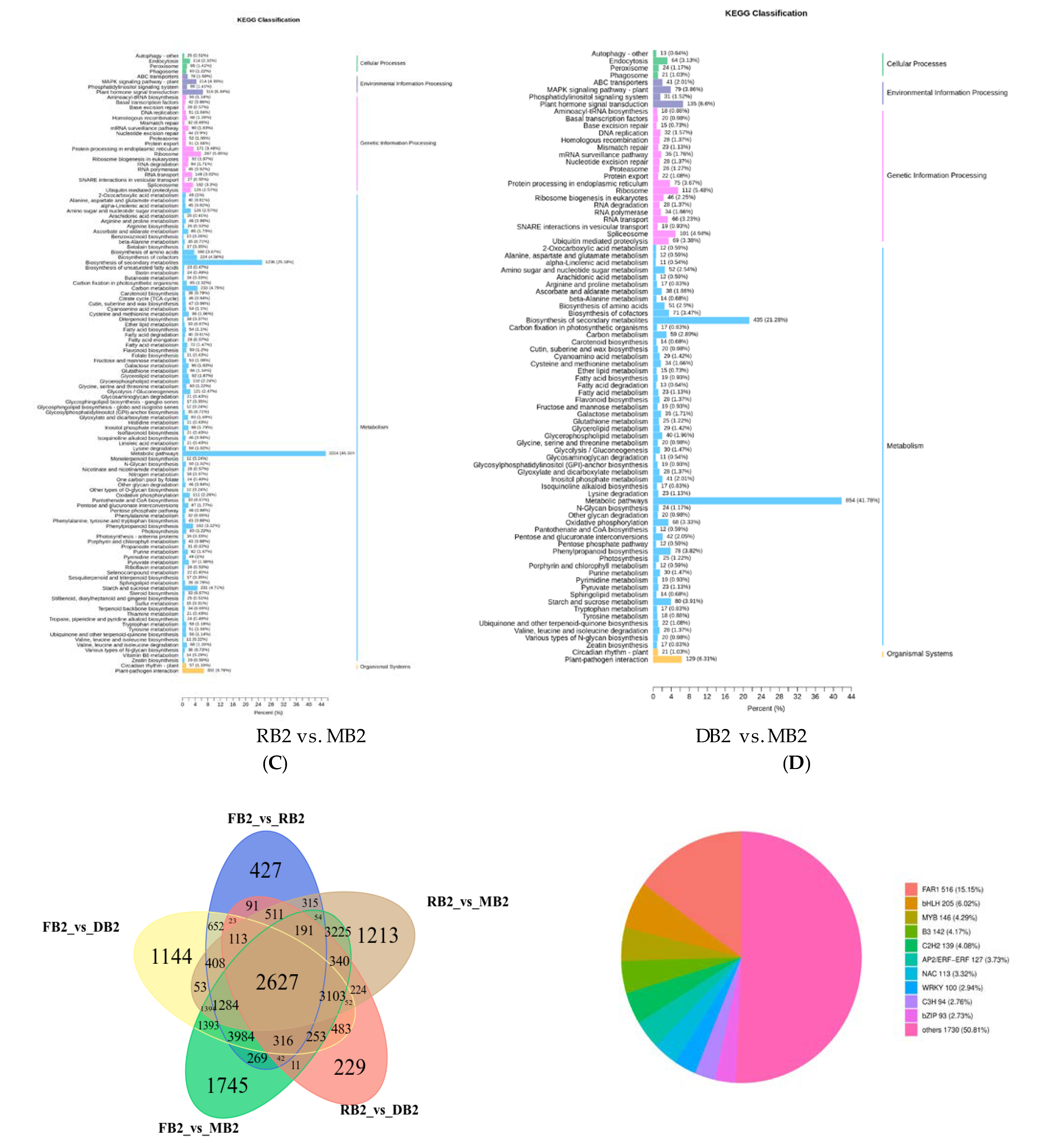
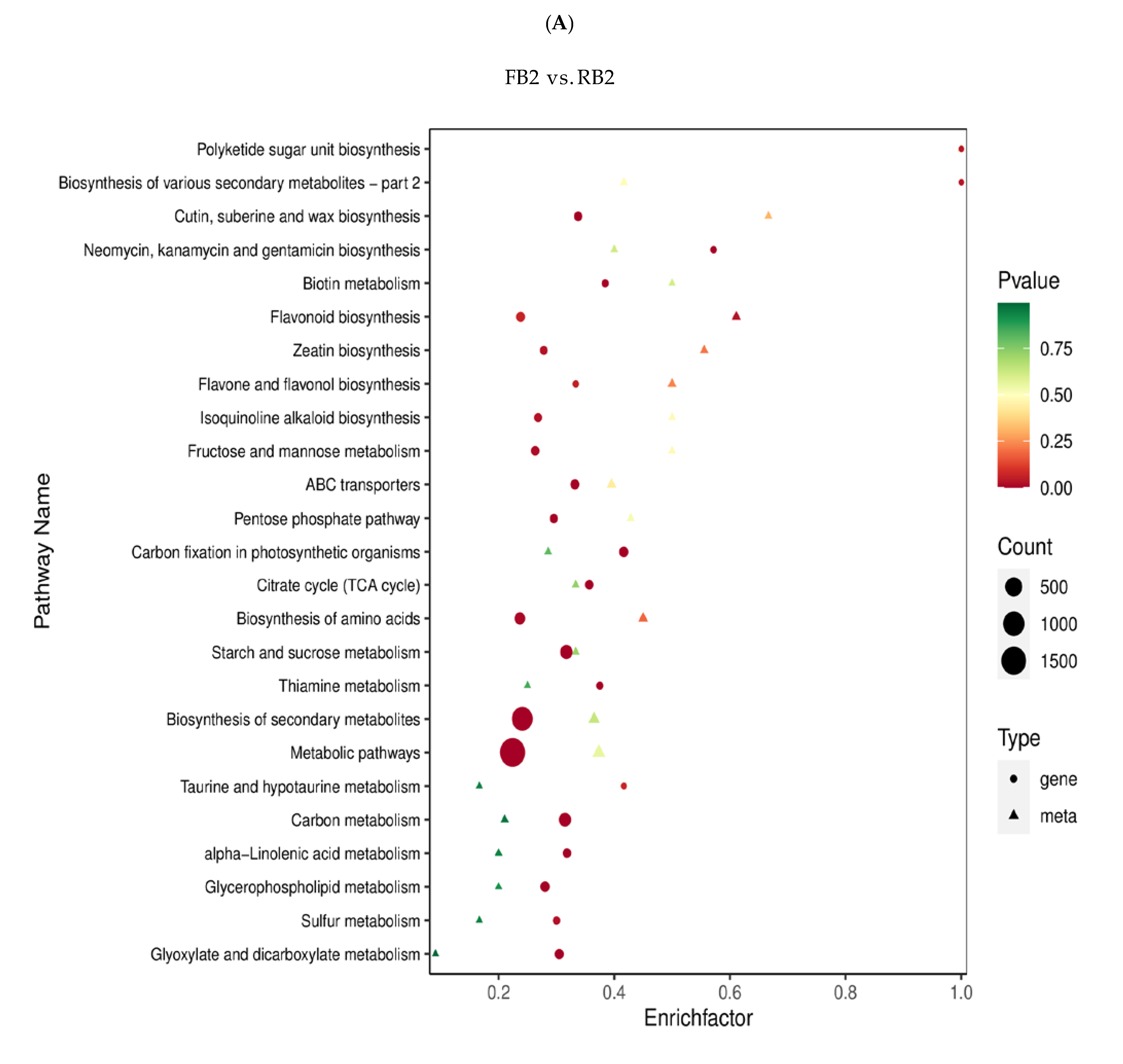
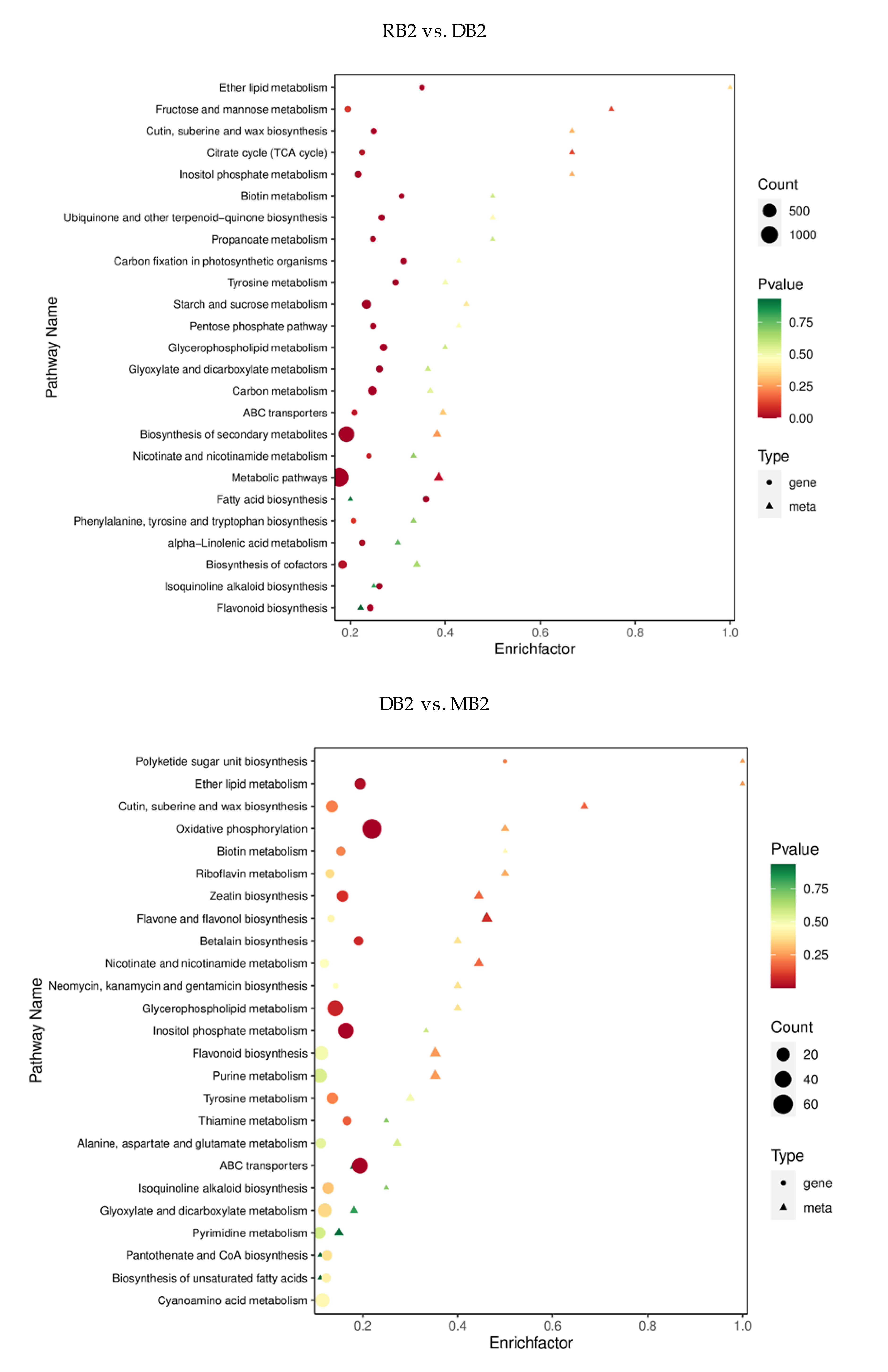
2.4. Functional Annotation and Enrichment Analysis of Differentially Expressed Genes
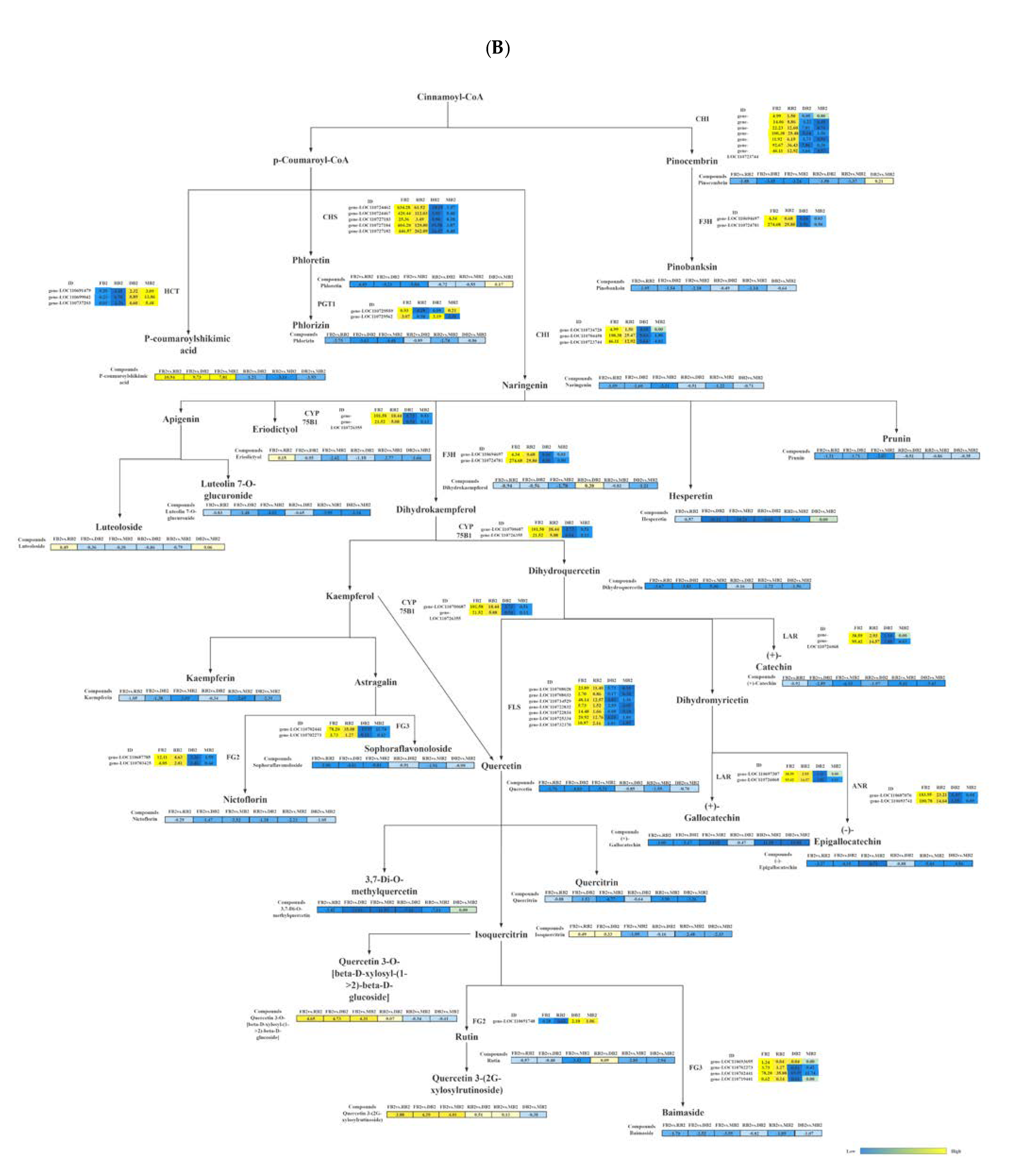
2.5. Combined Metabolomic and Transcriptomic Analysis of Flavonoid Regulation
3. Discussion
4. Materials and Methods
4.1. Materials and Sample Preparation
4.2. Extraction, Detection, and Qualitative and Quantitative Analysis of Metabolites
4.3. Transcriptome Sequencing and Data Analysis
4.4. Combined Transcriptome and Metabolome Analysis
4.5. RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escuredo, O.; González Martín, M.I.; Wells Moncada, G.; Fischer, S.; Hernández Hierro, J.M. Amino Acid Profile of the Quinoa (Chenopodium quinoa Willd.) Using near Infrared Spectroscopy and Chemometric Techniques. J. Cereal Sci. 2014, 60, 67–74. [Google Scholar] [CrossRef]
- Vega-Gálvez, A.; Miranda, M.; Vergara, J.; Uribe, E.; Puente, L.; Martínez, E.A. Nutrition Facts and Functional Potential of Quinoa (Chenopodium quinoa Willd.), an Ancient Andean Grain: A Review. J. Sci. Food Agric. 2010, 90, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Kozioł, M.J. Chemical Composition and Nutritional Evaluation of Quinoa (Chenopodium quinoa Willd.). J. Food Compos. Anal. 1992, 5, 35–68. [Google Scholar] [CrossRef]
- Abderrahim, F.; Huanatico, E.; Segura, R.; Arribas, S.; Gonzalez, M.C.; Condezo-Hoyos, L. Physical Features, Phenolic Compounds, Betalains and Total Antioxidant Capacity of Coloured Quinoa Seeds (Chenopodium quinoa Willd.) from Peruvian Altiplano. Food Chem. 2015, 183, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Lamothe, L.M.; Srichuwong, S.; Reuhs, B.L.; Hamaker, B.R. Quinoa (Chenopodium quinoa W.) and Amaranth (Amaranthus caudatus L.) Provide Dietary Fibres High in Pectic Substances and Xyloglucans. Food Chem. 2015, 167, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, X.; Zhang, B.; Chen, P.X.; Liu, R.; Tsao, R. Characterisation of Phenolics, Betanins and Antioxidant Activities in Seeds of Three Chenopodium quinoa Willd. Genotypes. Food Chem. 2015, 166, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, R.R.; Espinoza, C.; Jacobsne, S.E. Nutritional Value and Use of the Andean Crops Quinoa (Chenopodium quinoa) and Kaniwa (Chenopodium pallidicaule). Food Rev. Int. 2003, 19, 179–189. [Google Scholar] [CrossRef]
- Bhargava, A.; Shukla, S.; Ohri, D. Chenopodium quinoa–An Indian Perspective. Ind. Crops Prod. 2006, 23, 73–87. [Google Scholar] [CrossRef]
- Jacobsen, S.E. The Worldwide Potential for Quinoa (Chenopodium quinoa Willd.). Food Rev. Int. 2003, 19, 167–177. [Google Scholar] [CrossRef]
- FAO. Food and Agriculture Organization of the United States/World Health Organization/United Nations University, Energy and Protein Requirements; Report of a Joint FAO/WHO/UNU Meeting; World Health Organization: Geneva, Switzerland, 1985. [Google Scholar]
- Aloisi, I.; Parrotta, L.; Ruiz, K.B.; Landi, C.; Bini, L.; Cai, G.; Biondi, S.; Del Duca, S. New Insight into Quinoa Seed Quality Under Salinity: Changes in Proteomic and Amino Acid Profiles, Phenolic Content, and Antioxidant Activity of Protein Extracts. Front. Plant Sci. 2016, 7, 656. [Google Scholar] [CrossRef] [PubMed]
- Schijlen, E.G.W.; Ric de Vos, C.H.R.; Tunen, A.J.; Bovy, A.G. Modification of Flavonoid Biosynthesis in Crop Plants. Phytochemistry 2004, 65, 2631–2648. [Google Scholar] [CrossRef]
- Fowler, Z.L.; Koffas, M.A.G. Biosynthesis and Biotechnological Production of Flavanones: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2009, 83, 799–808. [Google Scholar] [CrossRef]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent Advances in the Transcriptional Regulation of the Flavonoid Biosynthetic Pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef]
- Gao, J.; Ren, R.; Wei, Y.; Jin, J.; Ahmad, S.; Lu, C.; Wu, J.; Zheng, C.; Yang, F.; Zhu, G. Comparative Metabolomic Analysis Reveals Distinct Flavonoid Biosynthesis Regulation for Leaf Color Development of Cymbidium sinense ‘Red Sun’. Int. J. Mol. Sci. 2020, 21, 1869. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lv, Q.; Ma, C.; Qu, J.; Cai, F.; Deng, J.; Huang, J.; Ran, P.; Shi, T.; Chen, Q. Metabolite Profiling and Transcriptome Analyses Provide Insights into the Flavonoid Biosynthesis in the Developing Seed of Tartary Buckwheat (Fagopyrum tataricum). J. Agric. Food Chem. 2019, 67, 11262–11276. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Huang, Z.; Lyu, L.; Li, W.; Wu, W. Integrative Analysis of the Metabolome and Transcriptome Provides Insights into the Mechanisms of Flavonoid Biosynthesis in Blackberry. Food Res. Int. 2022, 153, 110948. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zhao, L.; Liu, Y.; Zhang, A.; Xiao, S.; Dai, X.; Yuan, R.; Zhou, Z.; Cao, Q. Metabolomic and Transcriptomic Analyses of the Flavonoid Biosynthetic Pathway for the Accumulation of Anthocyanins and Other Flavonoids in Sweet potato Root Skin and Leaf Vein Base. J. Agric. Food Chem. 2022, 70, 2574–2588. [Google Scholar] [CrossRef]
- Ding, T.; Zhang, R.; Zhang, H.; Zhou, Z.; Liu, C.; Wu, M.; Wang, H.; Dong, H.; Liu, J.; Yao, J.L.; et al. Identification of Gene co-Expression Networks and Key Genes Regulating Flavonoid Accumulation in Apple (Malus × domestica) Fruit Skin. Plant Sci. 2021, 304, 110747. [Google Scholar] [CrossRef]
- Alvarez-Jubete, L.; Wijngaard, H.; Arendt, E.K.; Gallagher, E. Polyphenol Composition and In Vitro Antioxidant Activity of Amaranth, Quinoa Buckwheat and Wheat as Affected by Sprouting and Baking. Food Chem. 2010, 119, 770–778. [Google Scholar] [CrossRef]
- Loh, Y.C.; Chan, S.Y.; Tew, W.Y.; Oo, C.W.; Yam, M.F. New Flavonoid-Based Compound Synthesis Strategy for Antihypertensive Drug Development. Life Sci. 2020, 249, 117512. [Google Scholar] [CrossRef] [PubMed]
- Winkel-Shirley, B. It Takes a Garden. How Work on Diverse Plant Species Has Contributed to an Understanding of Flavonoid Metabolism. Plant Physiol. 2001, 127, 1399–1404. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Biosynthesis of Flavonoids and Effects of Stress. Curr. Opin. Plant Biol. 2002, 5, 218–223. [Google Scholar] [CrossRef]
- Harvaux, M.; Kloppstech, K. The Protective Functions of Carotenoid and Flavonoid Pigments Against Excess Visible Radiation at Chilling Temperature Investigated in Arabidopsis npq and tt Mutants. Planta 2001, 213, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Han, P.; Li, Y.; Wang, W.; Lai, D.; Zhou, L. Quinoa Secondary Metabolites and Their Biological Activities or Functions. Molecules 2019, 24, 2512. [Google Scholar] [CrossRef] [PubMed]
- Abugoch, L.E. Quinoa (Chenopodium quinoa Willd.) composition, chemistry, nutritional, and functional properties. Adv. Food Nutr. Res. 2009, 58, 1–31. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Pu, Z.; Wang, J.; Zheng, Y.; Li, Y.; Wei, Y. Regulation, evolution, and functionality of flavonoids in cereal crops. Biotechnol. Lett. 2013, 35, 1765–1780. [Google Scholar] [CrossRef]
- Liu, J. Quinoa. In Bioactive Factors and Processing Technology for Cereal Foods; Springer: Singapore, 2019; pp. 207–216. [Google Scholar] [CrossRef]
- Murphy, K.; Matanguihan, J. Quinoa: Improvement and Sustainable Production (Murphy/Quinoa: Improvement and Sustainable Production). In Nutritional Properties of Quinoa; Wiley: Hoboken, NJ, USA, 2015; pp. 1–18. [Google Scholar] [CrossRef]
- Ferrer, J.L.; Austin, M.B.; Stewart, C., Jr.; Noel, J.P. Structure and Function of Enzymes Involved in the Biosynthesis of Phenylpropanoids. Plant Physiol. Biochem. 2008, 46, 356–370. [Google Scholar] [CrossRef]
- Jiang, N.; Doseff, A.I.; Grotewold, E. Flavones: From Biosynthesis to Health Benefits. Plants 2016, 5, 27. [Google Scholar] [CrossRef]
- Han, Y.; Chi, J.; Zhang, M.; Zhang, R.; Fan, S.; Huang, F.; Xue, K.; Liu, L. Characterization of Saponins and Phenolic Compounds: Antioxidant Activity and Inhibitory Effects on α-Glucosidase in Different Varieties of Colored Quinoa (Chenopodium quinoa Willd.). Biosci. Biotechnol. Biochem. 2019, 83, 2128–2139. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Kong, Z.; Huan, X.; Li, L.; Zhang, P.; Wang, Q.; Guo, Y.; Zhu, W.; Qin, P. Transcriptomics and Metabolomics Analyses of the Mechanism of Flavonoid Synthesis in Seeds of Differently Colored Quinoa Strains. Genomics 2022, 114, 138–148. [Google Scholar] [CrossRef]
- Repo-Carrasco-Valencia, R.; Hellström, J.K.; Pihlava, J.M.; Mattila, P.H. Flavonoids and Other Phenolic Compounds in Andean Indigenous Grains: Quinoa (Chenopodium quinoa), Kañiwa (Chenopodium pallidicaule) and Kiwicha (Amaranthus caudatus). Food Chem. 2010, 120, 128–133. [Google Scholar] [CrossRef]
- Saleh, N.; Nozzolillo, C.; Altosaar, I. Flavonoid variations in Avena species. Biochem. Syst. Ecol. 1988, 16, 597–599. [Google Scholar] [CrossRef]
- Wu, P.; Liu, A.; Li, L.J. Metabolomics and transcriptome analysis of the biosynthesis mechanism of flavonoids in the seeds of Euryale ferox Salisb at different developmental stages. Mol. Genet. Genom. 2021, 296, 953–970. [Google Scholar] [CrossRef]
- Yilmaz, Y.; Toledo, R.T. Major Flavonoids in Grape Seeds and Skins: Antioxidant Capacity of Catechin, Epicatechin, and Gallic Acid. J. Agric. Food Chem. 2004, 52, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, U.A.; Joseph, C.M.; Phillips, A. Flavonoids Released Naturally fromAlfalfa SeedsEnhance GrowthRateofRhizobium meliloti. Plant Physiol. 1991, 98, 797–803. [Google Scholar] [CrossRef]
- Holton, T.A.; Cornish, E.C. Genetics and Biochemistry of Anthocyanin Biosynthesis. Plant Cell 1995, 7, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Guo, J.; Zhou, Q.; Xin, Y.; Wang, G.; Xu, L.A. De Novo Transcriptome Analysis Revealed Genes Involved in Flavonoid Biosynthesis, Transport and Regulation in Ginkgo biloba. Ind. Crops Prod. 2018, 124, 226–235. [Google Scholar] [CrossRef]
- Payne, C.T.; Zhang, F.; Lloyd, A.M. GL3 Encodes a bHLH Protein That Regulates Trichome Development in Arabidopsis Through Interaction with GL1 and TTG1. Genetics 2000, 156, 1349–1362. [Google Scholar] [CrossRef]
- Liu, J.; Osbourn, A.; Ma, P. MYB Transcription Factors as Regulators of Phenylpropanoid Metabolism in Plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Premathilake, A.T.; Ni, J.; Bai, S.; Tao, R.; Ahmad, M.; Teng, Y. R2R3-MYB Transcription Factor PpMYB17 Positively Regulates Flavonoid Biosynthesis in Pear Fruit. Planta 2020, 252, 59. [Google Scholar] [CrossRef]
- Huang, W.J.; Khaldun, A.B.M.; Chen, J.J.; Zhang, C.; Lv, H.; Yuan, L.; Wang, Y. A R2R3-MYB Transcription Factor Regulates the Flavonol Biosynthetic Pathway in a Traditional Chinese Medicinal Plant, Epimedium sagittatum. Front. Plant Sci. 2016, 7, 1089. [Google Scholar] [CrossRef]
- Xie, Y.; Tan, H.; Ma, Z.; Huang, J. DELLA Proteins Promote Anthocyanin Biosynthesis via Sequestering MYBL2 and JAZ Suppressors of the MYB/bHLH/WD40 Complex in Arabidopsis thaliana. Mol. Plant 2016, 9, 711–721. [Google Scholar] [CrossRef]
- Jaakola, L. New Insights into the Regulation of Anthocyanin Biosynthesis in Fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef]
- Liu, Y.; Tikunov, Y.; Schouten, R.E.; Marcelis, L.F.M.; Visser, R.G.F.; Bovy, A. Anthocyanin Biosynthesis and Degradation Mechanisms in Solanaceous Vegetables: A Review. Front. Chem. 2018, 6, 52. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A Novel Integrated Method for Large-Scale Detection, Identification, and Quantification of Widely Targeted Metabolites: Application in the Study of Rice Metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef]
- Eriksson, L.; Kettanehwold, N.; Trygg, J.; Wikström, C.; Wold, S. Multi-And Megavariate Data Analysis: Part I: Basic Principles and Applications, 8th ed.; Umetrics Inc.: San Jose, CA, USA, 2006; pp. 263–336. [Google Scholar] [CrossRef]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-Discovery Approach for Sample Matching of a Nerve-Agent Precursor Using Liquid Chromatography-Mass Spectrometry, XCMS, and Chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]
- An, Z.; Chen, Y.; Zhang, R.; Song, Y.; Sun, J.; He, J.; Bai, J.; Dong, L.L.; Zhan, Q.; Abliz, Z. Integrated Ionization Approach for RRLC-MS/MS-Based Metabonomics: Finding Potential Biomarkers for Lung Cancer. J. Proteome Res. 2010, 9, 4071–4081. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Xia, J. MetaboAnalystR: An R Package for Flexible and Reproducible Analysis of Metabolomics Data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef]
- Major, H.J.; Williams, R.; Wilson, A.J.; Wilson, I.D. A Metabonomic Analysis of Plasma from Zucker Rat Strains Using Gas Chromatography/Mass Spectrometry and Pattern Recognition. Rapid Commun. Mass Spectrom. 2006, 20, 3295–3302. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data Integration in Plant Biology: The O2PLS Method for Combined Modeling of Transcript and Metabolite Data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Li, M.; Zeng, Y.; Ge, L.; Gong, J.; Weng, C.; Yang, C.; Yang, J.; Fang, Y.; Li, Q.; Zou, T.; et al. Evaluation of the influences of low dose polybrominated diphenyl ethers exposure on human early retinal developent. Environ. Int. 2022, 163, 107187. [Google Scholar] [CrossRef]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.W. Evaluation of O2PLS in Omics Data Integration. BMC Bioinform. 2016, 17, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compared Samples | Total number of DEGs with Significant Difference | Total Number of DEGs Significantly Up-Regulated | Total Number of DEGs Significantly Down-Regulated |
|---|---|---|---|
| DB2_vs_MB2 | 6671 | 2638 | 4033 |
| FB2_vs_DB2 | 17,282 | 7986 | 9296 |
| FB2_vs_MB2 | 20,231 | 8398 | 11,833 |
| FB2_vs_RB2 | 11,307 | 5900 | 5407 |
| RB2_vs_DB2 | 8609 | 2916 | 5693 |
| RB2_vs_MB2 | 15,107 | 5957 | 9150 |
| KEGG Pathway | Gene Name | KEGG | Meta Name | Compounds | PCC |
|---|---|---|---|---|---|
| Flavonoid biosynthesis | Gene-LOC110704458 | K01859 [EC:5.5.1.6] | MWSHY0124 | Pinocembrin | 0.9025 |
| gene-LOC110723744 | K01859 [EC:5.5.1.6] | MWSHY0124 | Pinocembrin | 0.8990 | |
| gene-LOC110724781 | K00475 [EC:1.14.11.9] | mws0914 | Pinobanksin | 0.9714 | |
| gene-LOC110694697 | K00475 [EC:1.14.11.9] | mws0914 | Pinobanksin | 0.8201 | |
| gene-LOC110704458 | K01859 [EC:5.5.1.6] | pme0376 | Naringenin | 0.9580 | |
| gene-LOC110734728 | K01859 [EC:5.5.1.6] | pme0376 | Naringenin | 0.8333 | |
| gene-LOC110723744 | K01859 [EC:5.5.1.6] | pme0376 | Naringenin | 0.8695 | |
| gene-LOC110724462 | K00660[EC:2.3.1.74] | pme1201 | Phloretin | 0.8160 | |
| gene-LOC110727183 | K00660[EC:2.3.1.74] | pme1201 | Phloretin | 0.8081 | |
| gene-LOC110700687 | K05280 [EC:1.14.14.82] | mws0044 | Dihydroquercetin | 0.8936 | |
| gene-LOC110726355 | K05280 [EC:1.14.14.82] | mws0044 | Dihydroquercetin | 0.8943 | |
| gene-LOC110714529 | K05278 [EC:1.14.20.6] | pme2954 | Quercetin | 0.8902 | |
| gene-LOC110732370 | K05278 [EC:1.14.20.6] | pme2954 | Quercetin | 0.9283 | |
| gene-LOC110697307 | K13081 [EC:1.17.1.3] | mws0049 | Gallocatechin | 0.9966 | |
| gene-LOC110726068 | K13081 [EC:1.17.1.3] | mws0049 | Gallocatechin | 0.9270 | |
| gene-LOC110687076 | K08695 [EC:1.3.1.77] | mws0042 | Epigallocatechin | 0.9000 | |
| gene-LOC110693741 | K08695 [EC:1.3.1.77] | mws0042 | Epigallocatechin | 0.9052 | |
| Flavone and flavonol biosynthesis | gene-LOC110693695 | K22794 [EC:2.4.1.239 2.4.1.-] | Lmyn001269 | Sophoraflavonoloside | 0.8397 |
| gene-LOC110702273 | K22794 [EC:2.4.1.239 2.4.1.-] | Lmyn001269 | Sophoraflavonoloside | 0.8597 | |
| gene-LOC110702441 | K22794 [EC:2.4.1.239 2.4.1.-] | Lmyn001269 | Sophoraflavonoloside | 0.9534 | |
| gene-LOC110687785 | K22772 [EC:2.4.1.159] | MWSHY0050 | Nictoflorin | 0.8557 | |
| gene-LOC110703425 | K22772 [EC:2.4.1.159] | MWSHY0050 | Nictoflorin | 0.9468 | |
| gene-LOC110693695 | K22794 [EC:2.4.1.239 2.4.1.-] | MWSHY0162 | Baimaside | 0.9084 | |
| gene-LOC110702273 | K22794 [EC:2.4.1.239 2.4.1.-] | MWSHY0162 | Baimaside | 0.8454 | |
| gene-LOC110702441 | K22794 [EC:2.4.1.239 2.4.1.-] | MWSHY0162 | Baimaside | 0.9706 | |
| gene-LOC110719441 | K22794 [EC:2.4.1.239 2.4.1.-] | MWSHY0162 | Baimaside | 0.8579 | |
| gene-LOC110700687 | K05280 [EC:1.14.14.82] | pme2954 | Quercetin | 0.8817 | |
| gene-LOC110693695 | K22794 [EC:2.4.1.239 2.4.1.-] | pme2954 | Quercetin | 0.8290 | |
| gene-LOC110726355 | K05280 [EC:1.14.14.82] | pme2954 | Quercetin | 0.8807 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Yao, L.; Li, Q.; Xie, H.; Guo, Y.; Huang, T.; Zhang, X.; Liu, J.; Zhang, P.; Li, L.; et al. Integrative Analysis of the Metabolome and Transcriptome Provides Insights into the Mechanisms of Flavonoid Biosynthesis in Quinoa Seeds at Different Developmental Stages. Metabolites 2022, 12, 887. https://doi.org/10.3390/metabo12100887
Wang Q, Yao L, Li Q, Xie H, Guo Y, Huang T, Zhang X, Liu J, Zhang P, Li L, et al. Integrative Analysis of the Metabolome and Transcriptome Provides Insights into the Mechanisms of Flavonoid Biosynthesis in Quinoa Seeds at Different Developmental Stages. Metabolites. 2022; 12(10):887. https://doi.org/10.3390/metabo12100887
Chicago/Turabian StyleWang, Qianchao, Lan Yao, Qunying Li, Heng Xie, Yirui Guo, Tingzhi Huang, Xuesong Zhang, Junna Liu, Ping Zhang, Li Li, and et al. 2022. "Integrative Analysis of the Metabolome and Transcriptome Provides Insights into the Mechanisms of Flavonoid Biosynthesis in Quinoa Seeds at Different Developmental Stages" Metabolites 12, no. 10: 887. https://doi.org/10.3390/metabo12100887
APA StyleWang, Q., Yao, L., Li, Q., Xie, H., Guo, Y., Huang, T., Zhang, X., Liu, J., Zhang, P., Li, L., & Qin, P. (2022). Integrative Analysis of the Metabolome and Transcriptome Provides Insights into the Mechanisms of Flavonoid Biosynthesis in Quinoa Seeds at Different Developmental Stages. Metabolites, 12(10), 887. https://doi.org/10.3390/metabo12100887