
Metabolome Genome-Wide Association Study Identifies 74 Novel Genomic Regions Influencing Plasma Metabolites Levels

 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
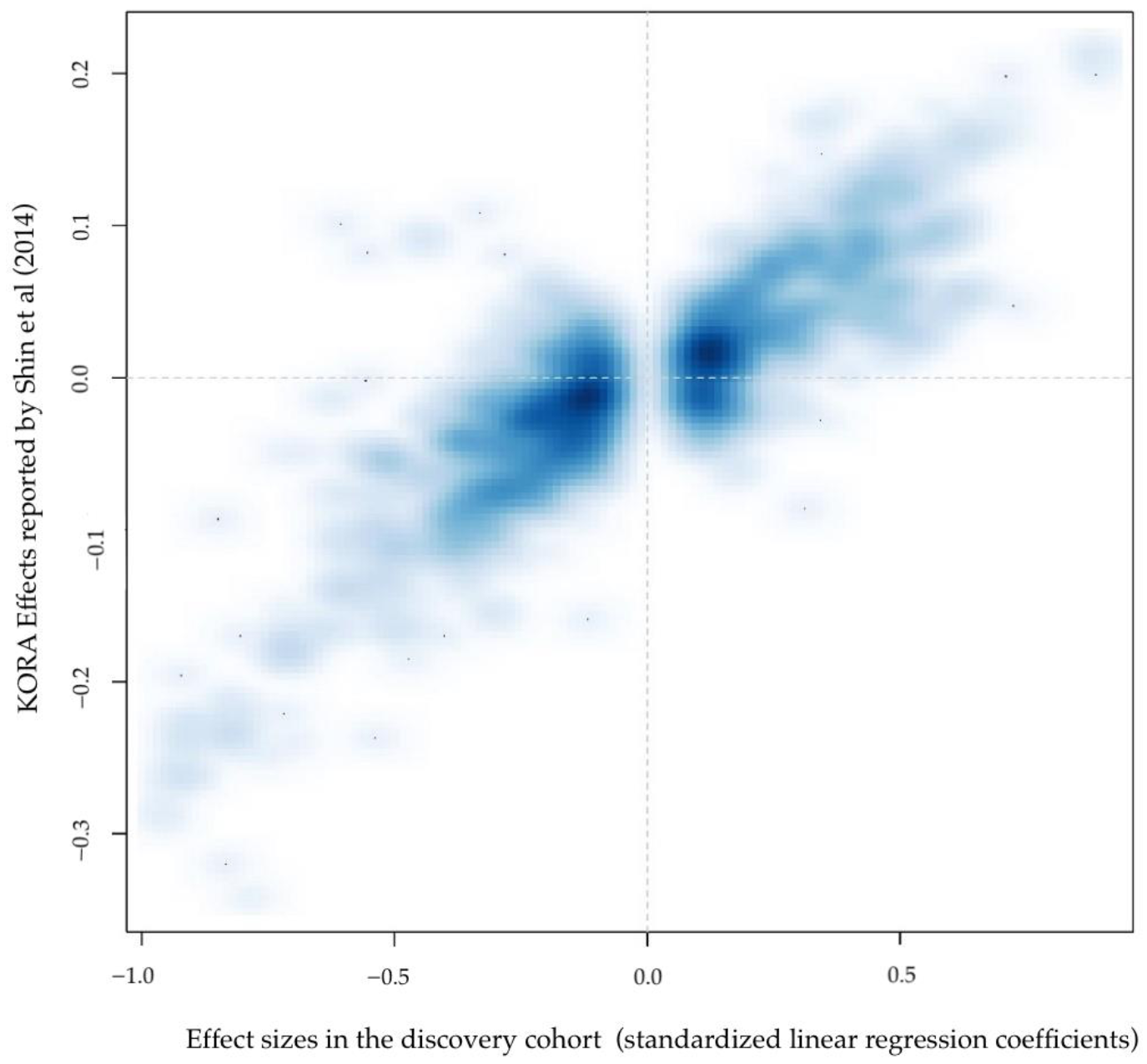
2. Results
3. Discussion
4. Materials and Methods
4.1. Metabolomic Profiling
4.2. Quality Control
4.3. Ultrahigh Performance Liquid Chromatography–Tandem Mass Spectroscopy (UPLC–MS/MS)
4.4. Bioinformatics
4.5. Data Extraction and Compound Identification
4.6. Data Quality
4.7. Metabolite Quantification and Data Normalization
4.8. Genotyping and Imputation
4.9. Kooperative Gesundheitsforschung in der Region Augsburg (KORA)
4.10. Statistical Analysis
4.11. Annotations
4.12. Meta-Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sebedio, J.L. Metabolomics, Nutrition, and Potential Biomarkers of Food Quality, Intake, and Health Status. Adv. Food Nutr. Res. 2017, 82, 83–116. [Google Scholar] [CrossRef] [PubMed]
- Pallister, T.; Sharafi, M.; Lachance, G.; Pirastu, N.; Mohney, R.P.; MacGregor, A.; Feskens, E.J.; Duffy, V.; Spector, T.D.; Menni, C. Food Preference Patterns in a UK Twin Cohort. Twin. Res. Hum. Genet. 2015, 18, 793–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, I.; Hankemeier, T.; Van der Graaf, P.H.; Knibbe, C.A.J.; Van Hasselt, J.G.C. Integrating clinical metabolomics-based biomarker discovery and clinical pharmacology to enable precision medicine. Eur. J. Pharm. Sci. 2017, 109, S15–S21. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.H.; Hauser, E.R.; Bain, J.R.; Muehlbauer, M.J.; Haynes, C.; Stevens, R.D.; Wenner, B.R.; Dowdy, Z.E.; Granger, C.B.; Ginsburg, G.S.; et al. High heritability of metabolomic profiles in families burdened with premature cardiovascular disease. Mol. Syst. Biol. 2009, 5, 258. [Google Scholar] [CrossRef]
- Demirkan, A.; Van Duijn, C.M.; Ugocsai, P.; Isaacs, A.; Pramstaller, P.P.; Liebisch, G.; Wilson, J.F.; Johansson, A.; Rudan, I.; Aulchenko, Y.S.; et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genet. 2012, 8, e1002490. [Google Scholar] [CrossRef] [Green Version]
- Draisma, H.H.M.; Pool, R.; Kobl, M.; Jansen, R.; Petersen, A.K.; Vaarhorst, A.A.M.; Yet, I.; Haller, T.; Demirkan, A.; Esko, T.; et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nat. Commun. 2015, 6, 7208. [Google Scholar] [CrossRef] [Green Version]
- Illig, T.; Gieger, C.; Zhai, G.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmuller, G.; Kato, B.S.; Mewes, H.W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Kettunen, J.; Tukiainen, T.; Sarin, A.P.; Ortega-Alonso, A.; Tikkanen, E.; Lyytikainen, L.P.; Kangas, A.J.; Soininen, P.; Wurtz, P.; Silander, K.; et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat. Genet. 2012, 44, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sekula, P.; Wuttke, M.; Wahrheit, J.; Hausknecht, B.; Schultheiss, U.T.; Gronwald, W.; Schlosser, P.; Tucci, S.; Ekici, A.B.; et al. Genome-Wide Association Studies of Metabolites in Patients with CKD Identify Multiple Loci and Illuminate Tubular Transport Mechanisms. J. Am. Soc. Nephrol. 2018, 29, 1513–1524. [Google Scholar] [CrossRef]
- Long, T.; Hicks, M.; Yu, H.C.; Biggs, W.H.; Kirkness, E.F.; Menni, C.; Zierer, J.; Small, K.S.; Mangino, M.; Messier, H.; et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 2017, 49, 568–578. [Google Scholar] [CrossRef]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Suhre, K.; Shin, S.Y.; Petersen, A.K.; Mohney, R.P.; Meredith, D.; Wägele, B.; Altmaier, E.; Deloukas, P.; Erdmann, J.; Grundberg, E.; et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011, 477, 54–60. [Google Scholar] [CrossRef]
- Lotta, L.A.; Pietzner, M.; Stewart, I.D.; Wittemans, L.B.L.; Li, C.; Bonelli, R.; Raffler, J.; Biggs, E.K.; Oliver-Williams, C.; Auyeung, V.P.W.; et al. A cross-platform approach identifies genetic regulators of human metabolism and health. Nat. Genet. 2021, 53, 54–64. [Google Scholar] [CrossRef]
- Raffler, J.; Friedrich, N.; Arnold, M.; Kacprowski, T.; Rueedi, R.; Altmaier, E.; Bergmann, S.; Budde, K.; Gieger, C.; Homuth, G.; et al. Genome-Wide Association Study with Targeted and Non-targeted NMR Metabolomics Identifies 15 Novel Loci of Urinary Human Metabolic Individuality. PLoS Genet. 2015, 11, e1005487. [Google Scholar] [CrossRef] [Green Version]
- Rueedi, R.; Ledda, M.; Nicholls, A.W.; Salek, R.M.; Marques-Vidal, P.; Morya, E.; Sameshima, K.; Montoliu, I.; Da Silva, L.; Collino, S.; et al. Genome-wide association study of metabolic traits reveals novel gene-metabolite-disease links. PLoS Genet. 2014, 10, e1004132. [Google Scholar] [CrossRef]
- Suhre, K.; Wallaschofski, H.; Raffler, J.; Friedrich, N.; Haring, R.; Michael, K.; Wasner, C.; Krebs, A.; Kronenberg, F.; Chang, D.; et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 2011, 43, 565–569. [Google Scholar] [CrossRef]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef]
- Nag, A.; Kurushima, Y.; Bowyer, R.C.E.; Wells, P.M.; Weiss, S.; Pietzner, M.; Kocher, T.; Raffler, J.; Völker, U.; Mangino, M.; et al. Genome-wide scan identifies novel genetic loci regulating salivary metabolite levels. Hum. Mol. Genet. 2020, 29, 864–875. [Google Scholar] [CrossRef]
- Devlin, B.; Roeder, K. Genomic control for association studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K.; et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Pe’er, I.; Yelensky, R.; Altshuler, D.; Daly, M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 2008, 32, 381–385. [Google Scholar] [CrossRef]
- Wood, A.R.; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.H.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; Kutalik, Z.; et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, C.B.; Kolvraa, S.; Kolvraa, A.; Stenbroen, V.; Kjeldsen, M.; Ensenauer, R.; Tein, I.; Matern, D.; Rinaldo, P.; Vianey-Saban, C.; et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum. Genet. 2008, 124, 43–56. [Google Scholar] [CrossRef]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Nie, S.K.; Chen, G.Q.; Cao, X.B.; Zhang, Y.J. Cerebrotendinous xanthomatosis: A comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2014, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Grohmann, K.; Lauffer, H.; Lauenstein, P.; Hoffmann, G.F.; Seidlitz, G. Hereditary Orotic Aciduria with Epilepsy and without Megaloblastic Anemia. Neuropediatrics 2015, 46, 123–125. [Google Scholar] [CrossRef]
- Saeed, A.; Floris, F.; Andersson, U.; Pikuleva, I.; Lovgren-Sandblom, A.; Bjerke, M.; Paucar, M.; Wallin, A.; Svenningsson, P.; Bjorkhem, I. 7alpha-hydroxy-3-oxo-4-cholestenoic acid in cerebrospinal fluid reflects the integrity of the blood-brain barrier. J. Lipid Res. 2014, 55, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Saeed, A.A.; Edstrom, E.; Pikuleva, I.; Eggertsen, G.; Bjorkhem, I. On the importance of albumin binding for the flux of 7alpha-hydroxy-3-oxo-4-cholestenoic acid in the brain. J. Lipid Res. 2017, 58, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Block, W.D.; Westhoff, M.H.; Steele, B.F. Histidine metabolism in the human adult: Histidine blood tolerance, and the effect of continued free L-histidine ingestion on the concentration of imidazole compounds in blood and urine. J. Nutr. 1967, 91, 189–194. [Google Scholar] [CrossRef] [PubMed]
- El-Batch, M.; Ibrahim, W.; Said, S. Effect of histidine on autotaxin activity in experimentally induced liver fibrosis. J. Biochem. Mol. Toxicol. 2011, 25, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.L.; Wu, S.T.; Yin, M.C.; Chen, H.T.; Chen, H.C. Protective effects from carnosine and histidine on acetaminophen-induced liver injury. J. Food Sci. 2009, 74, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Keys, H.R.; Cantor, J.R.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Freinkman, E.; Schweitzer, L.D.; Sabatini, D.M. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 2018, 559, 632–636. [Google Scholar] [CrossRef]
- Adkins, D.E.; McClay, J.L.; Vunck, S.A.; Batman, A.M.; Vann, R.E.; Clark, S.L.; Souza, R.P.; Crowley, J.J.; Sullivan, P.F.; Van den Oord, E.J.C.G.; et al. Behavioral metabolomics analysis identifies novel neurochemical signatures in methamphetamine sensitization. Genes Brain Behav. 2013, 12, 780–791. [Google Scholar] [CrossRef]
- Hong, H.; Fill, T.; Leadlay, P.F. A Common Origin for Guanidinobutanoate Starter Units in Antifungal Natural Products. Angew. Chem. Int. Edit. 2013, 52, 13096–13099. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat Genet 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Loh, P.O.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Delaneau, O.; Howie, B.; Cox, A.J.; Zagury, J.F.; Marchini, J. Haplotype estimation using sequencing reads. Am J Hum Genet 2013, 93, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Wichmann, H.E.; Gieger, C.; Illig, T.; for the MONICA/KORA Study Group. KORA-gen-resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen 2005, 67 (Suppl. 1), S26–S30. [Google Scholar] [CrossRef] [Green Version]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium; Duncan, L.; et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ferreira, T.; Morris, A.P.; Medland, S.E.; GIANT Consortium; DIAGRAM Consortium; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012, 44, 369–375. [Google Scholar] [CrossRef]
- McKusick, V.A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hysi, P.G.; Mangino, M.; Christofidou, P.; Falchi, M.; Karoly, E.D.; NIHR Bioresource Investigators; Mohney, R.P.; Valdes, A.M.; Spector, T.D.; Menni, C. Metabolome Genome-Wide Association Study Identifies 74 Novel Genomic Regions Influencing Plasma Metabolites Levels. Metabolites 2022, 12, 61. https://doi.org/10.3390/metabo12010061
Hysi PG, Mangino M, Christofidou P, Falchi M, Karoly ED, NIHR Bioresource Investigators, Mohney RP, Valdes AM, Spector TD, Menni C. Metabolome Genome-Wide Association Study Identifies 74 Novel Genomic Regions Influencing Plasma Metabolites Levels. Metabolites. 2022; 12(1):61. https://doi.org/10.3390/metabo12010061
Chicago/Turabian StyleHysi, Pirro G., Massimo Mangino, Paraskevi Christofidou, Mario Falchi, Edward D. Karoly, NIHR Bioresource Investigators, Robert P. Mohney, Ana M. Valdes, Tim D. Spector, and Cristina Menni. 2022. "Metabolome Genome-Wide Association Study Identifies 74 Novel Genomic Regions Influencing Plasma Metabolites Levels" Metabolites 12, no. 1: 61. https://doi.org/10.3390/metabo12010061
APA StyleHysi, P. G., Mangino, M., Christofidou, P., Falchi, M., Karoly, E. D., NIHR Bioresource Investigators, Mohney, R. P., Valdes, A. M., Spector, T. D., & Menni, C. (2022). Metabolome Genome-Wide Association Study Identifies 74 Novel Genomic Regions Influencing Plasma Metabolites Levels. Metabolites, 12(1), 61. https://doi.org/10.3390/metabo12010061