
A Mediation Approach to Discovering Causal Relationships between the Metabolome and DNA Methylation in Type 1 Diabetes

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Study Design and Participants
4.2. DNA Methylation
4.3. Metabolomics
4.4. Statistical Analysis
4.5. Biological Interpretation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rakyan, V.K.; Beyan, H.; Down, T.A.; Hawa, M.I.; Maslau, S.; Aden, D.; Daunay, A.; Busato, F.; Mein, C.; Manfras, B.; et al. Identification of Type 1 Diabetes-Associated DNA Methylation Variable Positions That Precede Disease Diagnosis. PLoS Genet. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Tuomilehto, J. The Emerging Global Epidemic of Type 1 Diabetes. Curr. Diabetes Rep. 2013, 13, 795–804. [Google Scholar] [CrossRef]
- Polychronakos, C.; Li, Q. Understanding type 1 diabetes through genetics: Advances and prospects. Nat. Rev. Genet. 2011, 12, 781–792. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; A Erlich, H.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, M.; Zhang, W.; Concepcion, E.; Yi, Z.; Tomer, Y. DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J. Autoimmun. 2014, 50, 33–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.K.; Vanderlinden, L.A.; Dong, F.; Carry, P.M.; Seifert, J.; Waugh, K.; Shorrosh, H.; Fingerlin, T.; Frohnert, B.I.; Yang, I.V.; et al. Longitudinal DNA methylation differences precede type 1 diabetes. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Orešič, M.; Simell, S.; Sysi-Aho, M.; Näntö-Salonen, K.; Seppänen-Laakso, T.; Parikka, V.; Katajamaa, M.; Hekkala, A.; Mattila, I.; Keskinen, P.; et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008, 205, 2975–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohnert, B.I.; Rewers, M.J. Metabolomics in childhood diabetes. Pediatr. Diabetes 2016, 17, 3–14. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Piunti, A.; Pasini, D. Epigenetic methylations and their connections with metabolism. Cell. Mol. Life Sci. 2013, 70, 1495–1508. [Google Scholar] [CrossRef]
- Putluri, N.; Shojaie, A.; Vasu, V.; Nalluri, S.; Vareed, S.K.; Putluri, V.; Vivekanandan-Giri, A.; Byun, J.; Pennathur, S.; Sana, T.R.; et al. Metabolomic Profiling Reveals a Role for Androgen in Activating Amino Acid Metabolism and Methylation in Prostate Cancer Cells. PLoS ONE 2011, 6, e21417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Wick, W.; Von Deimling, A. Isocitrate dehydrogenase mutations: A challenge to traditional views on the genesis and malignant progression of gliomas. Glia 2011, 59, 1200–1204. [Google Scholar] [CrossRef]
- Alakwaa, F.M.; Savelieff, M. Bioinformatics Analysis of Metabolomics Data Unveils Association of Metabolic Signatures with Methylation in Breast Cancer. J. Proteome Res. 2020, 19, 2879–2889. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, J.; Fang, D.; Lv, L.; Wu, W.; Shi, D.; Li, Y.; Yang, L.; Bian, X.; Wu, J.; et al. Multi-omic profiling reveals associations between the gut mucosal microbiome, the metabolome, and host DNA methylation associated gene expression in patients with colorectal cancer. BMC Microbiol. 2020, 20, 83. [Google Scholar] [CrossRef]
- Huang, Y.; Hui, Q.; I Walker, D.; Uppal, K.; Goldberg, J.; Jones, D.P.; Vaccarino, V.; Sun, Y.V. Untargeted metabolomics reveals multiple metabolites influencing smoking-related DNA methylation. Epigenomics 2018, 10, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Zaghlool, S.B.; O Mook-Kanamori, D.; Kader, S.; Stephan, N.; Halama, A.; Engelke, R.; Sarwath, H.; Al-Dous, E.K.; A Mohamoud, Y.; Roemisch-Margl, W.; et al. Deep molecular phenotypes link complex disorders and physiological insult to CpG methylation. Hum. Mol. Genet. 2018, 27, 1106–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanderWeele, T.J. Explanation in Causal Inference: Methods for Mediation and Interaction; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Fiske, S.T.; Gilbert, D.T.; Lindzey, G.; Jongsma, A.E. Handbook of Social Psychology, 5th ed.; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Johnson, R.K.; Vanderlinden, L.; DeFelice, B.C.; Kechris, K.; Uusitalo, U.; Fiehn, O.; Sontag, M.; Crume, T.; Beyerlein, A.; Lernmark, Å.; et al. Metabolite-related dietary patterns and the development of islet autoimmunity. Sci. Rep. 2019, 9, 14819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.K.; Vanderlinden, L.A.; DeFelice, B.C.; Uusitalo, U.; Seifert, J.; Fan, S.; Crume, T.; Fiehn, O.; Rewers, M.; Kechris, K.; et al. Metabolomics-related nutrient patterns at seroconversion and risk of progression to type 1 diabetes. Pediatr. Diabetes 2020, 21, 1202–1209. [Google Scholar] [CrossRef]
- Dutta, T.; Kudva, Y.C.; Persson, X.-M.T.; Schenck, L.A.; Ford, G.C.; Singh, R.J.; Carter, R.; Nair, K.S. Impact of Long-Term Poor and Good Glycemic Control on Metabolomics Alterations in Type 1 Diabetic People. J. Clin. Endocrinol. Metab. 2016, 101, 1023–1033. [Google Scholar] [CrossRef]
- Alkan, M.; Machavoine, F.; Rignault, R.; Dam, J.; Dy, M.; Thieblemont, N. Histidine Decarboxylase Deficiency Prevents Autoimmune Diabetes in NOD Mice. J. Diabetes Res. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Manca, M.L.; Solini, A.; Haukka, J.K.; Sandholm, N.; Forsblom, C.; Groop, P.-H.; Ferrannini, E. Differential metabolomic signatures of declining renal function in Types 1 and 2 diabetes. Nephrol. Dial. Transplant. 2020. [Google Scholar] [CrossRef]
- Husain, Z.; Kelly, M.A.; Eisenbarth, G.S.; Pugliese, A.; Awdeh, Z.L.; Larsen, C.; Alper, C.A. The MHC type 1 diabetes susceptibility gene is centromeric to HLA-DQB1. J. Autoimmun. 2008, 30, 266–272. [Google Scholar] [CrossRef]
- Gillberg, L.; Perfilyev, A.; Brøns, C.; Thomasen, M.; Grunnet, L.G.; Volkov, P.; Rosqvist, F.; Iggman, D.; Dahlman, I.; Risérus, U.; et al. Adipose tissue transcriptomics and epigenomics in low birthweight men and controls: Role of high-fat overfeeding. Diabetologia 2016, 59, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Rangasamy, S.; Naymik, M.; Monickaraj, F.; Legendre, C.; Balak, C.; Duggan, D.; Schork, N.; McGuire, P. Novel Genetic Variants in Extreme Phenotypes of Diabetic Retinopathy: DRGen Study. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1911. [Google Scholar]
- Errera, F.I.; Canani, L.H.; Yeh, E.; Kague, E.; Armelin-Correa, L.M.; Suzuki, O.T.; Tschiedel, B.; Silva, M.E.R.; Sertie, A.; Passos-Bueno, M.R.; et al. COL18A1 is highly expressed during human adipocyte differentiation and the SNP c.1136C > T in its "frizzled" motif is associated with obesity in diabetes type 2 patients. An. Acad. Bras. Ciências 2008, 80, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; Solanes, G.; Fernández-Alvarez, J.; Pujol, A.; Ferrer, J.; Asins, G.; Gomis, R.; Bosch, F. Expression of GLUT-2 antisense RNA in beta cells of transgenic mice leads to diabetes. J. Biol. Chem. 1994, 269, 28543–28546. [Google Scholar] [CrossRef]
- Nokoff, N.; Rewers, M. Pathogenesis of type 1 diabetes: Lessons from natural history studies of high-risk individuals. Ann. N. Y. Acad. Sci. 2013, 1281, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senderek, J.; Bergmann, C.; Stendel, C.; Kirfel, J.; Verpoorten, N.; De Jonghe, P.; Timmerman, V.; Chrast, R.; Verheijen, M.H.; Lemke, G.; et al. Mutations in a Gene Encoding a Novel SH3/TPR Domain Protein Cause Autosomal Recessive Charcot-Marie-Tooth Type 4C Neuropathy. Am. J. Hum. Genet. 2003, 73, 1106–1119. [Google Scholar] [CrossRef] [Green Version]
- Carry, P.M.; Vanderlinden, L.A.; Johnson, R.K.; Buckner, T.; Fiehn, O.; Steck, A.K.; Kechris, K.; Yang, I.; Fingerlin, T.E.; Rewers, M.; et al. Phospholipid Levels at Seroconversion Are Associated with Resolution of Persistent Islet Autoimmunity: The Diabetes Autoimmunity Study in the Young. Diabetes 2021, 70, 1592–1601. [Google Scholar] [CrossRef]
- Kavishwar, A.; Moore, A. Sphingomyelin Patches on Pancreatic Beta-cells Are Indicative of Insulin Secretory Capacity. J. Histochem. Cytochem. 2013, 61, 910–919. [Google Scholar] [CrossRef]
- Barlovic, D.P.; Harjutsalo, V.; Sandholm, N.; Forsblom, C.; Groop, P.-H.; on behalf of the FinnDiane Study Group. Sphingomyelin and progression of renal and coronary heart disease in individuals with type 1 diabetes. Diabetologia 2020, 63, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, V.-P.; Tynkkynen, T.; Soininen, P.; Forsblom, C.; Peltola, T.; Kangas, A.; Groop, P.-H.; Ala-Korpela, M. Sphingomyelin is associated with kidney disease in type 1 diabetes (The FinnDiane Study). Metabolomics 2012, 8, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofte, N.; Suvitaival, T.; Ahonen, L.; Winther, S.A.; Theilade, S.; Frimodt-Møller, M.; Ahluwalia, T.S.; Rossing, P. Lipidomic analysis reveals sphingomyelin and phosphatidylcholine species associated with renal impairment and all-cause mortality in type 1 diabetes. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Liu, G.-M.; Zeng, H.-D.; Zhang, C.-Y.; Xu, J.-W. Key genes associated with diabetes mellitus and hepatocellular carcinoma. Pathol. Res. Pr. 2019, 215, 152510. [Google Scholar] [CrossRef]
- Jones, D.P.; Park, Y.; Ziegler, T.R. Nutritional Metabolomics: Progress in Addressing Complexity in Diet and Health. Annu. Rev. Nutr. 2012, 32, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Rudra, P.; Shi, W.J.; Russell, P.; Vestal, B.; Tabakoff, B.; Hoffman, P.; Kechris, K.; Saba, L. Predictive modeling of miRNA-mediated predisposition to alcohol-related phenotypes in mouse. BMC Genom. 2018, 19, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rewers, M.; Norris, J.M.; Eisenbarth, G.S.; Erlich, H.A.; Beatya, B.; Klingensmithb, G.; Hoffmana, M.; Yub, L.; Bugawan, T.L.; Blairc, A.; et al. Beta-Cell Autoantibodies in Infants and Toddlers without IDDM Relatives: Diabetes Autoimmunity Study in the Young (DAISY). J. Autoimmun. 1996, 9, 405–410. [Google Scholar] [CrossRef]
- Rewers, M.; Bugawan, T.L.; Norris, J.M.; Blair, A.; Beaty, B.; Hoffman, M.; McDuffie, R.S.; Hamman, R.F.; Klingensmith, G.; Eisenbarth, G.S.; et al. Newborn screening for HLA markers associated with IDDM: Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia 1996, 39, 807–812. [Google Scholar] [CrossRef]
- Zhou, W.; Triche, T.J.; Laird, P.W.; Shen, H. SeSAMe: Reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018, 46, e123. [Google Scholar] [CrossRef] [Green Version]
- Skogerson, K.; Wohlgemuth, G.; Barupal, D.K.; Fiehn, O. The volatile compound BinBase mass spectral database. BMC Bioinform. 2011, 12, 321. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.S.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [Green Version]
- DeFelice, B.C.; Mehta, S.S.; Samra, S.; Čajka, T.; Wancewicz, B.; Fahrmann, J.F.; Fiehn, O. Mass Spectral Feature List Optimizer (MS-FLO): A Tool to Minimize False Positive Peak Reports in Untargeted Liquid Chromatography–Mass Spectroscopy (LC-MS) Data Processing. Anal. Chem. 2017, 89, 3250–3255. [Google Scholar] [CrossRef]
- Fan, S.; Kind, T.; Cajka, T.; Hazen, S.L.; Tang, W.H.W.; Kaddurah-Daouk, R.; Irvin, M.R.; Arnett, D.K.; Barupal, D.K.; Fiehn, O. Systematic Error Removal Using Random Forest for Normalizing Large-Scale Untargeted Lipidomics Data. Anal. Chem. 2019, 91, 3590–3596. [Google Scholar] [CrossRef]
- Box, G.E.P.; Cox, D.R. An Analysis of Transformations. J. R. Stat. Soc. Ser. B 1964, 26, 211–243. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; Global Biodiversity Information Facility: Copenhagen, Denmark, 2021. [Google Scholar]
- Yoshida, K.; Li, Y. Regmedint: Regression-Based Causal Mediation Analysis with an Interaction Term; R Package Version 0.2.1. 2020. Available online: https://CRAN.R-project.org/package=regmedint (accessed on 6 August 2021).
- Valeri, L.; VanderWeele, T.J. Mediation analysis allowing for exposure–mediator interactions and causal interpretation: Theoretical assumptions and implementation with SAS and SPSS macros. Psychol. Methods 2013, 18, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Yekutieli, D. False Discovery Rate–Adjusted Multiple Confidence Intervals for Selected Parameters. J. Am. Stat. Assoc. 2005, 100, 71–81. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Phipson, B.; Maksimovic, J.; Oshlack, A. missMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 2016, 32, 286–288. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.-T.; Guha, R.; Jadhav, A.; et al. The NCATS BioPlanet—An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef] [Green Version]
- Bonder, M.J.; Luijk, R.; Zhernakova, D.; Moed, M.; Deelen, P.; Vermaat, M.; Van Iterson, M.M.; Van Dijk, F.; Van Galen, M.; Bot, J.; et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat. Genet. 2017, 49, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.K.; Kilaru, V.; Kocak, M.; Almli, L.M.; Mercer, K.B.; Ressler, K.J.; A Tylavsky, F.; Conneely, K.N. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genom. 2014, 15, 145. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinformatics 2016, 54. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wen, Z.; Chen, S.; Bibiko, H.; Lowe, W. GeneBook: Multi-Types Gene ID Converting/Annotating; R Package Version 1.0. 2019. Available online: https://CRAN.R-project.org/package=GeneBook (accessed on 6 August 2021).
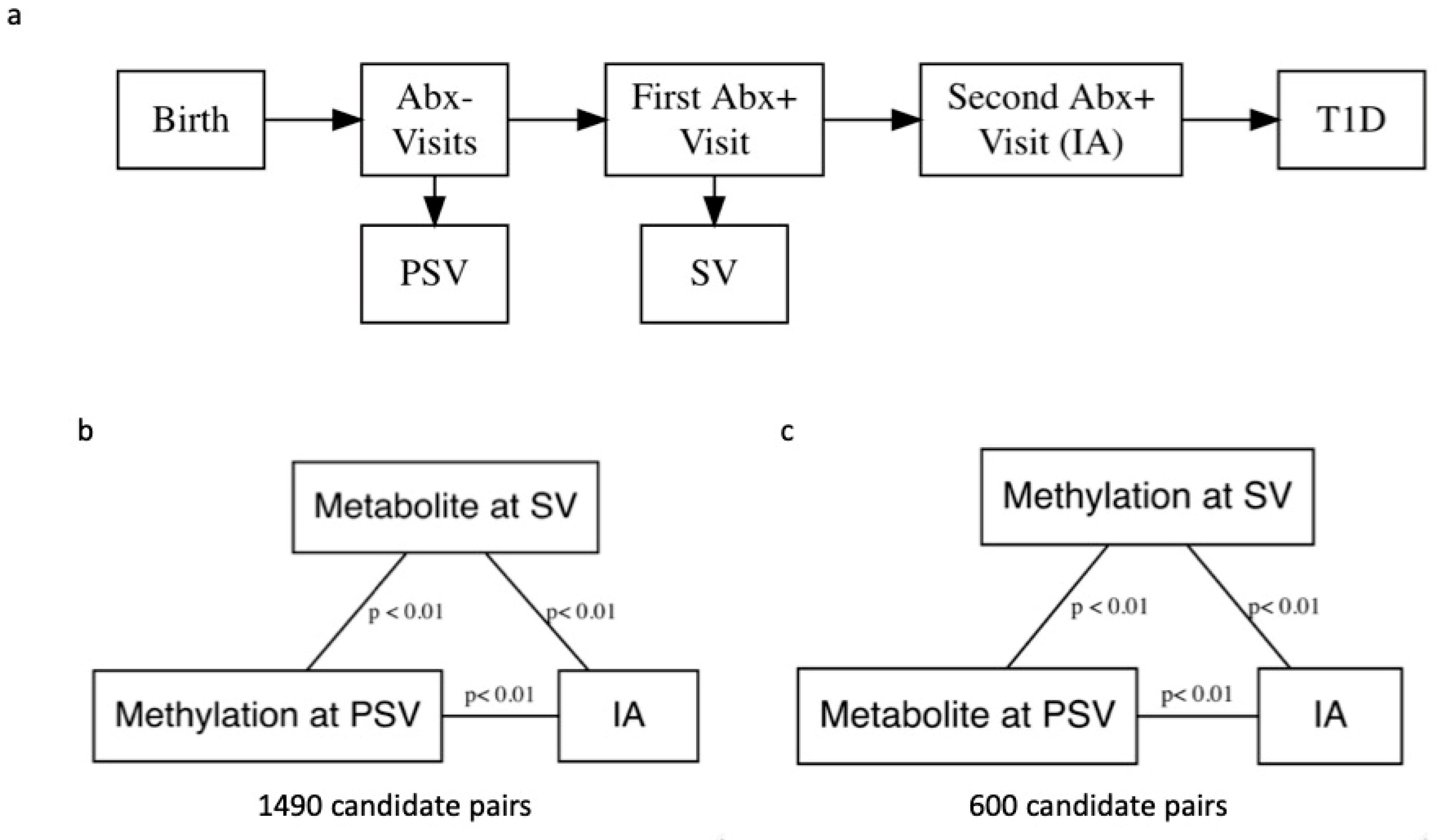

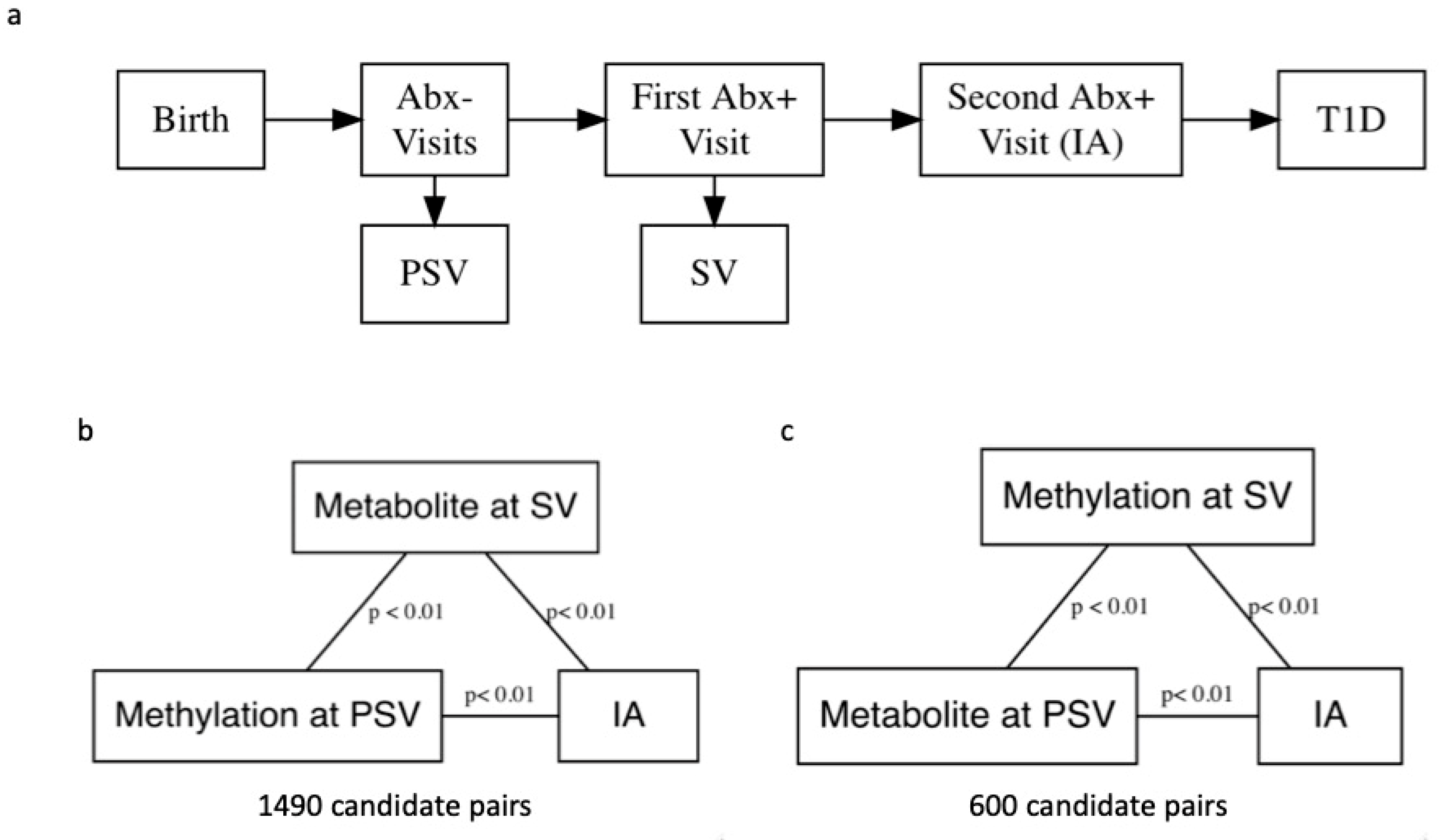{kind=link}
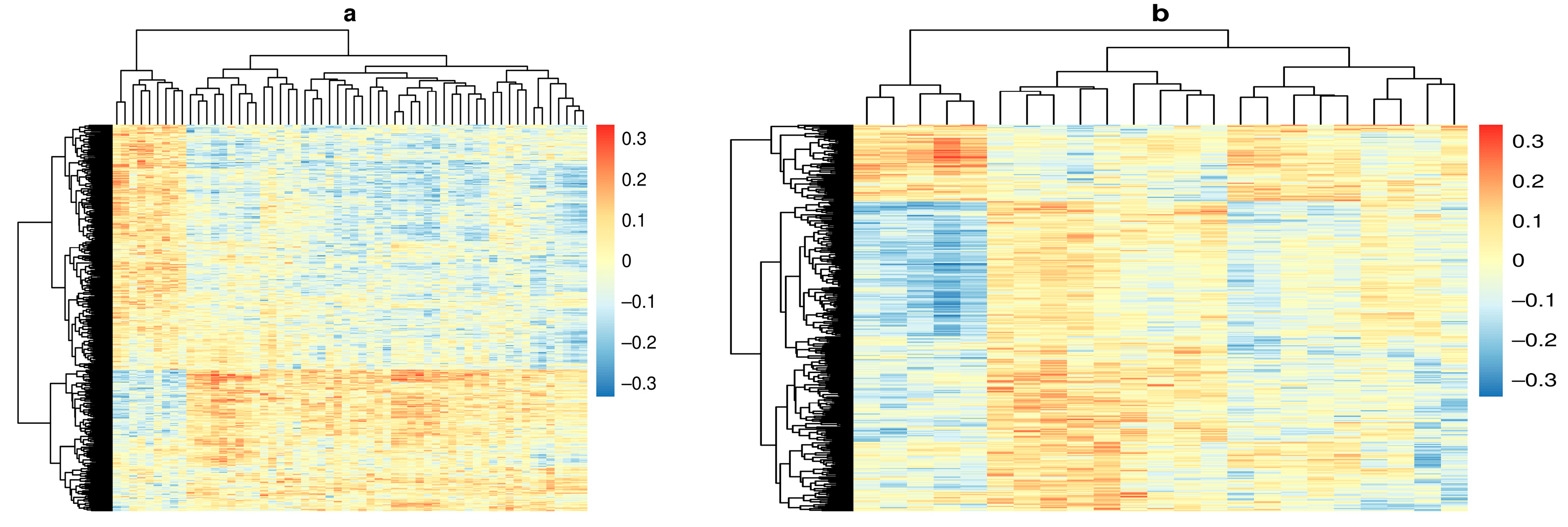
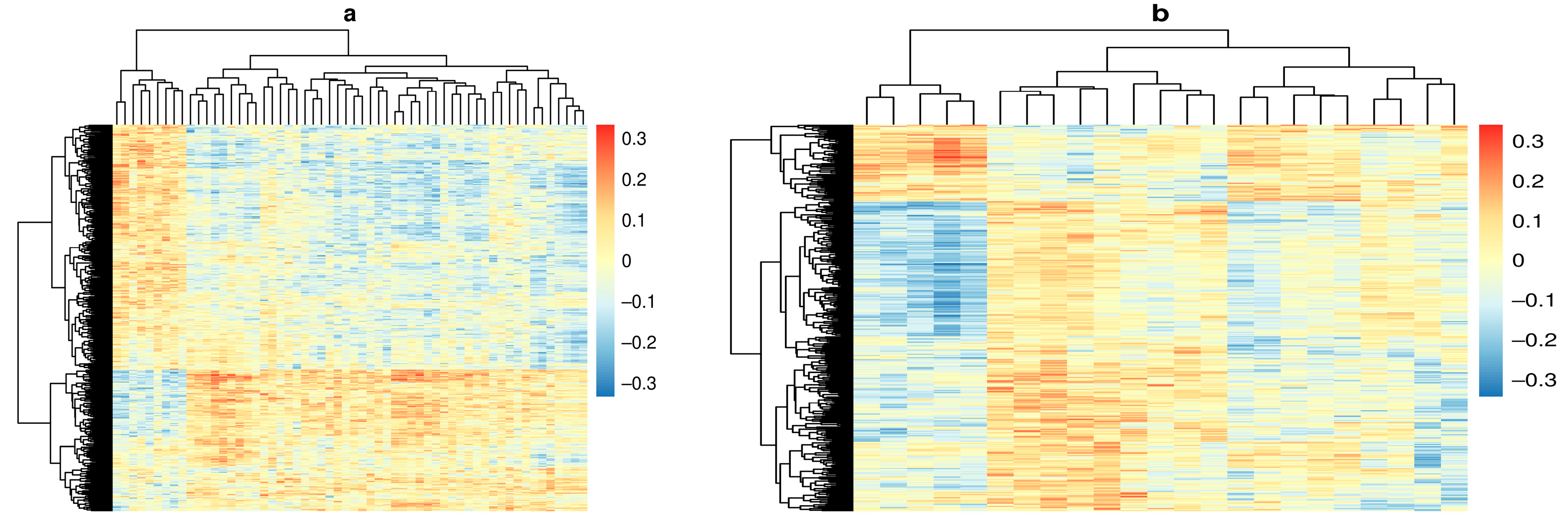{kind=link}
| Case (n = 92) | Control (n = 91) | Total (n = 183) | p Value | |
|---|---|---|---|---|
| Age | 0.387 1 | |||
| Mean (SD) | 6.2 (4.3) | 6.8 (4.2) | 6.5 (4.3) | |
| Range | 0.7–18.3 | 0.7–20.3 | 0.7–20.3 | |
| Non-Hispanic White | 0.756 2 | |||
| No | 22 (23.9%) | 20 (22.0%) | 42 (23.0%) | |
| Yes | 70 (76.1%) | 71 (78.0%) | 141 (77.0%) | |
| DR3/4 | 0.172 2 | |||
| No | 67 (72.8%) | 74 (81.3%) | 141 (77.0%) | |
| Yes | 25 (27.2%) | 17 (18.7%) | 42 (23.0%) | |
| Sex | 0.599 2 | |||
| Female | 44 (47.8%) | 40 (44.0%) | 84 (45.9%) | |
| Male | 48 (52.2%) | 51 (56.0%) | 99 (54.1%) | |
| FDR Status | 0.819 2 | |||
| First-degree relative with T1D | 49 (53.3%) | 50 (54.9%) | 99 (54.1%) | |
| General population (no first degree relative with T1D) | 43 (46.7%) | 41 (45.1%) | 84 (45.9%) |
| Metabolite | Probe | Position | Relation to Island | Gene | Type | Description | NIE | NIE CI Low | NIE CI High |
|---|---|---|---|---|---|---|---|---|---|
| Histidine | cg15688253 | chr1:1096717 | N_Shore | 0.766 | 0.418 | 0.993 | |||
| Histidine | cg15052330 | chr2:72360243 | OpenSea | CYP26B1 | 0.714 | 0.339 | 0.950 | ||
| PC (p-32:0) or PC (o-32:1) | cg01604946 | chr5:148398804 | OpenSea | SH3TC2 | Protein Coding | SH3 Domain; Tetratricopeptide Repeats 2; SH3TC2 Divergent Transcript | 0.663 | 0.342 | 0.895 |
| Histidine | cg19939773 | chr6:32729876 | Island | HLA-DQB2 | 0.785 | 0.439 | 0.994 | ||
| SM (d34:2) [M+HAc-H]- & [M+Cl]- _YLWSJLLZUHSIEA-CKSUKHGVSA-N | cg00390143 | chr12:132842539 | Island | GALNT9 | Protein Coding | Polypeptide N-Acetylgalactosaminyltransferase 9 | 0.777 | 0.494 | 0.999 |
| Histidine | cg01172082 | chr14:104645732 | Island | KIF26A | Protein Coding | Kinesin Family Member 26A | 0.759 | 0.390 | 0.989 |
| Histidine | cg07964219 | chr21:46847898 | S_Shore | COL18A1 | Protein Coding | Collagen Type XVIII Alpha 1 Chain; COL18A1 Antisense RNA 1; COL18A1 Antisense RNA 2 | 0.795 | 0.447 | 0.986 |
| Term | Number of Genes in Top 100 Probes | Number of Genes in Reference List | Expected | Fold Enrichment | p Value | FDR q Value |
|---|---|---|---|---|---|---|
| Formation of the cornified envelope (R-HSA-6809371) | 3 | 105 | 0.32 | 9.24 | 0.004 | 0.071 |
| Inositol phosphate metabolism (R-HSA-1483249) | 2 | 43 | 0.13 | 15.04 | 0.008 | 0.071 |
| Ion transport by P-type ATPases (R-HSA-936837) | 2 | 49 | 0.15 | 13.2 | 0.011 | 0.071 |
| Keratinization (R-HSA-6805567) | 3 | 159 | 0.49 | 6.1 | 0.014 | 0.071 |
| Transport of small molecules (R-HSA-382551) | 6 | 668 | 2.07 | 2.91 | 0.017 | 0.071 |
| Plasma lipoprotein assembly, remodeling, and clearance (R-HSA-174824) | 2 | 63 | 0.19 | 10.27 | 0.017 | 0.071 |
| Term | Number of Genes in Top 100 Probes | Number of Genes in Reference List | p Value | FDR q Value | Fold Enrichment | Genes |
|---|---|---|---|---|---|---|
| Interleukin receptor SHC signaling | 2 | 28 | 0.0031816 | 0.2497991 | 26.411141 | IL3;IL5RA |
| Ion transport by P-type ATPases | 2 | 36 | 0.0052218 | 0.2497991 | 20.188641 | ATP4B;ATP2B2 |
| Interleukin-3 signaling pathway | 2 | 45 | 0.0080651 | 0.2497991 | 15.955894 | IL3;IL5RA |
| Interleukin-3, interleukin-5, and GM-CSF signaling | 2 | 45 | 0.0080651 | 0.2497991 | 15.955894 | IL3;IL5RA |
| Regulation of NFAT transcription factors | 2 | 47 | 0.0087727 | 0.2497991 | 15.245211 | IL3;IKZF1 |
| Ion channel transport | 2 | 61 | 0.0144583 | 0.2497991 | 11.619521 | ATP4B;ATP2B2 |
| Sodium-coupled sulphate, di- and tri-carboxylate transporters | 1 | 5 | 0.0149116 | 0.2497991 | 84.474576 | SLC13A2 |
| Cytochrome P450 metabolism of vitamins | 1 | 6 | 0.0178676 | 0.2497991 | 67.576271 | CYP26B1 |
| Phase I of biological oxidations: non-cytochrome P450 enzymes | 1 | 7 | 0.0208149 | 0.2497991 | 56.310735 | LIPA |
| Eosinophils in the chemokine network of allergy | 1 | 8 | 0.0237535 | 0.2497991 | 48.263922 | IL3 |
| Small cell lung cancer | 2 | 84 | 0.0263601 | 0.2497991 | 8.350715 | LAMA2;TRAF5 |
| Hematopoietic cell lineage | 2 | 88 | 0.0287269 | 0.2497991 | 7.960706 | IL3;IL5RA |
| Transmembrane transport of small molecules | 4 | 432 | 0.0405993 | 0.2497991 | 3.256342 | ATP4B;SLC13A2;STEAP3;ATP2B2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vigers, T.; Vanderlinden, L.A.; Johnson, R.K.; Carry, P.M.; Yang, I.; DeFelice, B.C.; Kaizer, A.M.; Pyle, L.; Rewers, M.; Fiehn, O.; et al. A Mediation Approach to Discovering Causal Relationships between the Metabolome and DNA Methylation in Type 1 Diabetes. Metabolites 2021, 11, 542. https://doi.org/10.3390/metabo11080542
Vigers T, Vanderlinden LA, Johnson RK, Carry PM, Yang I, DeFelice BC, Kaizer AM, Pyle L, Rewers M, Fiehn O, et al. A Mediation Approach to Discovering Causal Relationships between the Metabolome and DNA Methylation in Type 1 Diabetes. Metabolites. 2021; 11(8):542. https://doi.org/10.3390/metabo11080542
Chicago/Turabian StyleVigers, Tim, Lauren A. Vanderlinden, Randi K. Johnson, Patrick M. Carry, Ivana Yang, Brian C. DeFelice, Alexander M. Kaizer, Laura Pyle, Marian Rewers, Oliver Fiehn, and et al. 2021. "A Mediation Approach to Discovering Causal Relationships between the Metabolome and DNA Methylation in Type 1 Diabetes" Metabolites 11, no. 8: 542. https://doi.org/10.3390/metabo11080542
APA StyleVigers, T., Vanderlinden, L. A., Johnson, R. K., Carry, P. M., Yang, I., DeFelice, B. C., Kaizer, A. M., Pyle, L., Rewers, M., Fiehn, O., Norris, J. M., & Kechris, K. (2021). A Mediation Approach to Discovering Causal Relationships between the Metabolome and DNA Methylation in Type 1 Diabetes. Metabolites, 11(8), 542. https://doi.org/10.3390/metabo11080542