
Combining HPLC-DAD-QTOF-MS and HPLC-SPE-NMR to Monitor In Vitro Vitetrifolin D Phase I and II Metabolism



Abstract
:1. Introduction
2. Results
2.1. HPLC-DAD-QTOF-MS Analysis
2.2. HPLC-DAD-QTOF-MS-Based Metabolite Characterization
2.3. Metabolite Identification
2.3.1. Single-Fold Oxidation
2.3.2. Twofold Oxidation
2.3.3. Threefold Oxidation
2.3.4. Hydrolysis
2.3.5. Glucuronidation
2.3.6. Sulfation
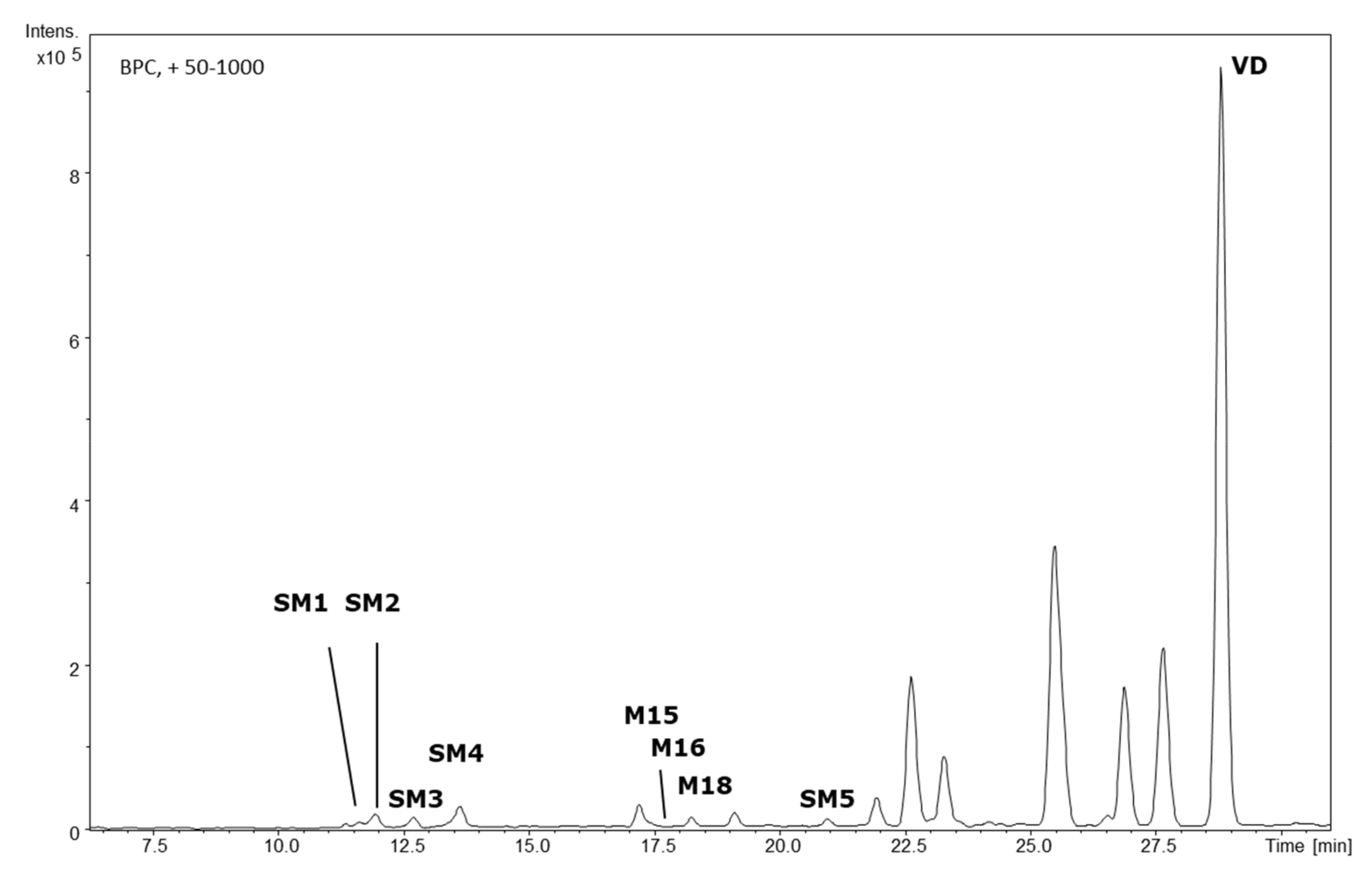
2.4. HPLC-SPE-NMR Analysis
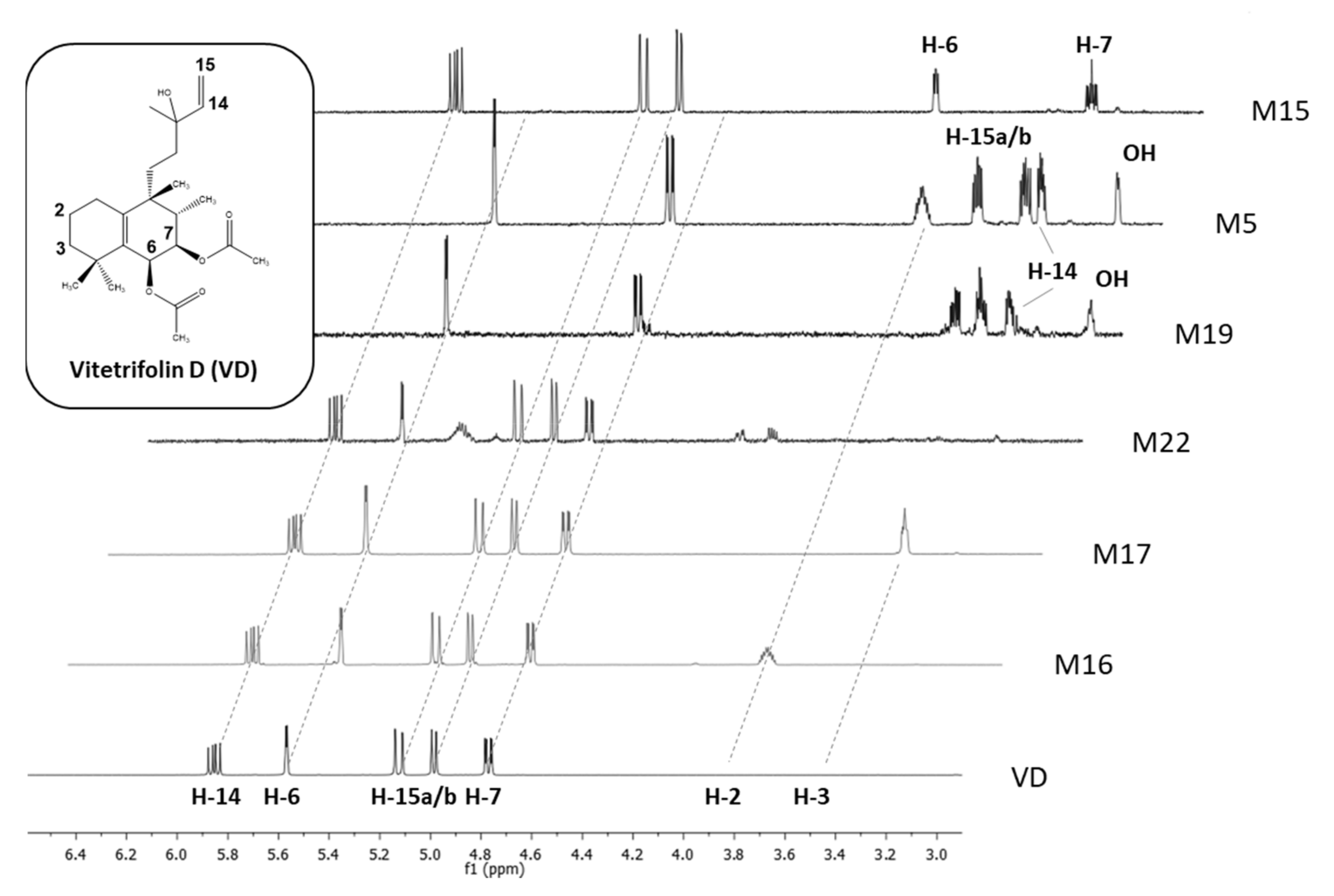
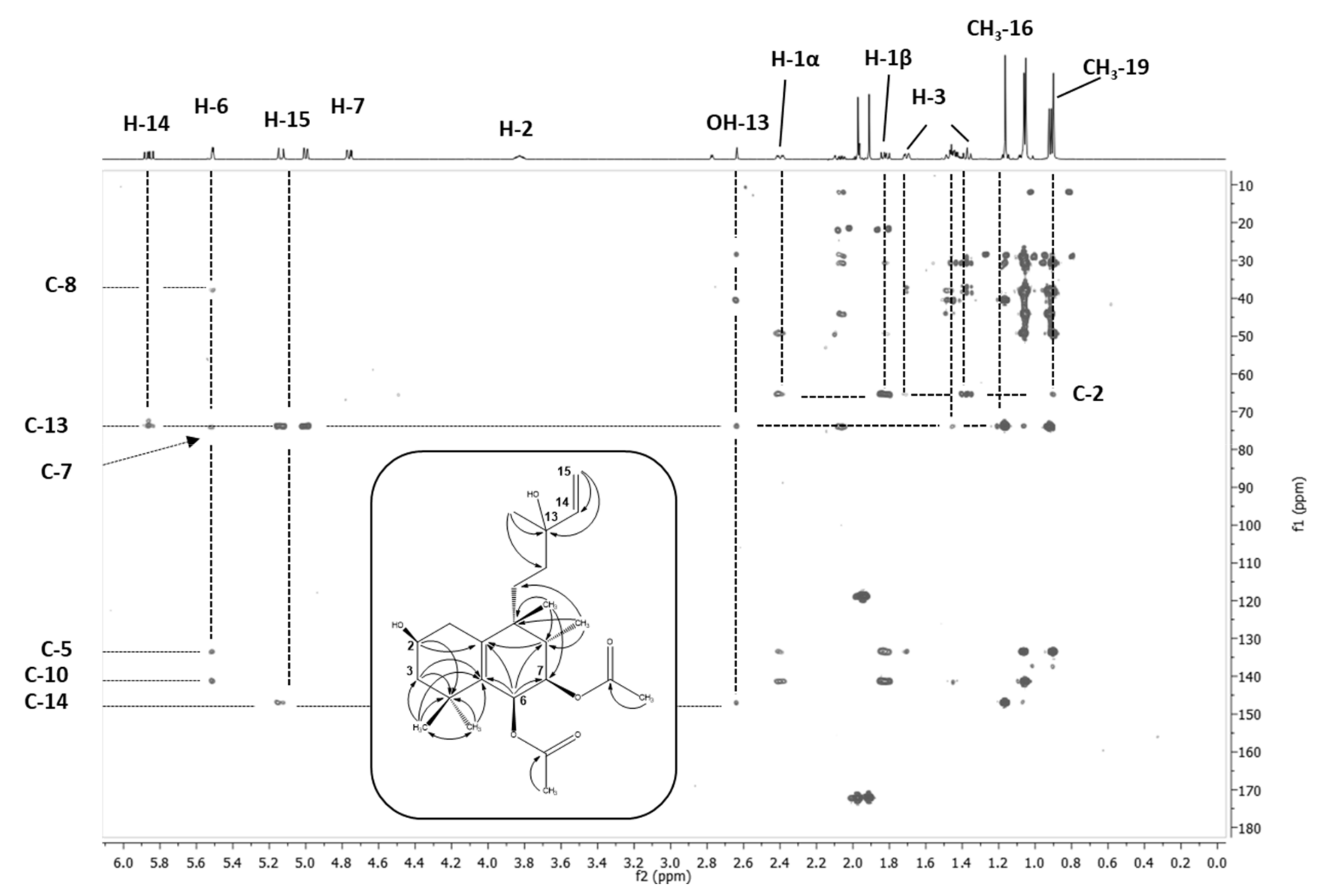
2.5. NMR-Based Structure Elucidation
2.5.1. Vitetrifolin D (VD)
2.5.2. Metabolites with Onefold Oxidation (M16, M17, M20, M22)
2.5.3. Metabolites with Twofold Oxidation (M18, M19)
2.5.4. Metabolites with Threefold Oxidation (M2, M5)
2.5.5. Metabolites with Hydrolysis of Acetate Groups (M15)
2.6. Analytes without NMR-Based Structural Characterization
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of the Vititrifolin D Stock Solution
4.3. Cellular Assay Incubation Conditions
4.3.1. HLM Phase I Metabolism Assay
4.3.2. S9 Fraction UDP-Glucuronosyltransferase (UGT) Assay
4.3.3. S9 Cell Fraction Sulfation Incubations
4.4. HPLC-DAD-QTOF-MS
4.5. HPLC-SPE-NMR
4.5.1. Instrumentation
4.5.2. Sample Preparation
4.5.3. HPLC-SPE-NMR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Caldwell, J.; Gardner, I.; Swales, N. An introduction to drug disposition: The basic principle of absorption, distribution, metabolism, and excretion. Toxicol. Pathol. 1995, 23, 102–112. [Google Scholar] [CrossRef]
- Roffey, S.J.; Obach, R.S.; Gedge, J.I.; Smith, D.A. What is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabelled drugs. Drug Metab. Rev. 2007, 39, 17–43. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kirchmair, J. Cheminformatics in natural product-based drug discovery. Mol. Inform. 2020, 39, e2000171. [Google Scholar] [CrossRef] [PubMed]
- Marathe, P.H.; Shyu, W.C.; Humphreys, W.G. The use of radiolabeled compounds for ADME studies in discovery and exploratory development. Curr. Pharm. Des. 2004, 10, 2991–3008. [Google Scholar] [CrossRef]
- Atzrodt, J.; Allen, J. Synthesis of radiolabeled compounds for clinical studies. In Drug Discovery and Evaluation: Methods in Clinical Pharmacology; Vogel, H.G., Maas, J., Gebauer, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 105–118. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Thiengsusuk, A.; Boonprasert, K.; Na-Bangchang, K. A systematic review of drug metabolism studies of plants with anticancer properties: Approaches applied and limitations. Eur. J. Drug Metab. Pharmacokinet. 2020, 45, 173–225. [Google Scholar] [CrossRef]
- Cho, H.J.; Yoon, I.S. Pharmacokinetic interactions of herbs with cytochrome p450 and p-glycoprotein. Evid. Based Complement. Alternat. Med. 2015, 2015, 736431. [Google Scholar] [CrossRef]
- Basheer, L.; Kerem, Z. Interactions between CYP3A4 and dietary polyphenols. Oxid. Med. Cell. Longev. 2015, 2015, 854015. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H.; Chin, Y.W. Multifaceted factors causing conflicting outcomes in herb-drug interactions. Pharmaceutics 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Auxtero, M.D.; Chalante, S.; Abade, M.R.; Jorge, R.; Fernandes, A.I. Potential herb-drug interactions in the management of age-related cognitive dysfunction. Pharmaceutics 2021, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Chugh, N.A.; Bali, S.; Koul, A. Integration of botanicals in contemporary medicine: Road blocks, checkpoints and go-ahead signals. Integr. Med. Res. 2018, 7, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.K.; Song, I.S. Pharmacokinetic drug-drug interactions and herb-drug interactions. Pharmaceutics 2021, 13, 610. [Google Scholar] [CrossRef] [PubMed]
- Haiyu, X.; Yanqiong, Z.; Ping, W.; Junhong, Z.; Hong, C.; Luoqi, Z.; Xia, D.; Chunhui, Z.; Dan, W.; Feng, L.; et al. A comprehensive review of integrative pharmacology-based investigation: A paradigm shift in traditional Chinese medicine. Acta Pharm. Sin. B 2021, in press. [Google Scholar] [CrossRef]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Ladumor, M.K.; Tiwari, S.; Patil, A.; Bhavsar, K.; Jhajra, S.; Prasad, B.; Singh, S. High-resolution mass spectrometry in metabolite identification. In Comprehensive Analytical Chemistry; Perez, S., Eichhorn, P., Barcelo, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 71, pp. 199–229. [Google Scholar] [CrossRef]
- Prasad, B.; Garg, A.; Takwani, H.; Singh, S. Metabolite identification by liquid chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2011, 30, 360–387. [Google Scholar] [CrossRef]
- Burns, D.C.; Reynolds, W.F. Minimizing the risk of deducing wrong natural product structures from NMR data. Magn. Reson. Chem. 2021, 59, 500–533. [Google Scholar] [CrossRef]
- Seger, C. Nuclear magnetic resonance of small molecules in natural products. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; Wiley: New York, NY, USA, 2014; pp. 1–12. [Google Scholar] [CrossRef]
- Seger, C.; Sturm, S.; Stuppner, H. Mass spectrometry and NMR spectroscopy: Modern high-end detectors for high resolution separation techniques—state of the art in natural product HPLC-MS, HPLC-NMR, and CE-MS hyphenations. Nat. Prod. Rep. 2013, 30, 970–987. [Google Scholar] [CrossRef]
- Sturm, S.; Seger, C. Liquid chromatography-nuclear magnetic resonance coupling as alternative to liquid chromatography-mass spectrometry hyphenations: Curious option or powerful and complementary routine tool? J. Chromatogr. A 2012, 1259, 50–61. [Google Scholar] [CrossRef]
- Gathungu, R.M.; Kautz, R.; Kristal, B.S.; Bird, S.S.; Vouros, P. The integration of LC-MS and NMR for the analysis of low molecular weight trace analytes in complex matrices. Mass Spectrom. Rev. 2020, 39, 35–54. [Google Scholar] [CrossRef]
- Van Die, M.D.; Burger, H.G.; Teede, H.J.; Bone, K.M. Vitex agnus-castus extracts for female reproductive disorders: A systematic review of clinical trials. Planta Med. 2013, 79, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Hoberg, E.; Orjala, J.; Meier, B.; Sticher, O. Diterpenoids from the fruits of Vitex agnus-castus. Phytochemistry 1999, 52, 1555–1558. [Google Scholar] [CrossRef]
- Meier, B.; Berger, D.; Hoberg, E.; Sticher, O.; Schaffner, W. Pharmacological activities of Vitex agnus-castus extracts in vitro. Phytomedicine 2000, 7, 373–381. [Google Scholar] [CrossRef]
- Jarry, H.; Leonhardt, S.; Gorkow, C.; Wuttke, W. In vitro prolactin but no LH and FSH release is inhibited by compounds in extracts of Agnus-castus: Direct evidence for a dopaminergic principle by the dopamine receptor assay. Exp. Clin. Endocrinol. 1994, 102, 448–454. [Google Scholar] [CrossRef]
- Yao, J.L.; Fang, S.M.; Liu, R.; Oppong, M.B.; Liu, E.W.; Fan, G.W.; Zhang, H. A Review on the terpenes from genus Vitex. Molecules 2016, 21, 1179. [Google Scholar] [CrossRef] [Green Version]
- Duffus, J.H.; Nordberg, M.; Templeton, D.M. Glossary of terms used in toxicology, 2nd edition (IUPAC Recommendations 2007). Pure Appl. Chem. 2007, 79, 1153–1344. [Google Scholar] [CrossRef] [Green Version]
- Suiko, M.; Kurogi, K.; Hashiguchi, T.; Sakakibara, Y.; Liu, M.C. Updated perspectives on the cytosolic sulfotransferases (SULTs) and SULT-mediated sulfation. Biosci. Biotechnol. Biochem. 2017, 81, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asha, S.; Vidyavathi, M. Role of human liver microsomes in in vitro metabolism of drugs—A review. Appl Biochem. Biotechnol. 2010, 160, 1699–1722. [Google Scholar] [CrossRef] [PubMed]
- Knights, K.M.; Stresser, D.M.; Miners, J.O.; Crespi, C.L. In vitro drug metabolism using liver microsomes. Curr. Protoc. Pharmacol. 2016, 74, 7.8.1–7.8.24. [Google Scholar] [CrossRef] [PubMed]
- Sturm, S.; Seger, C.; Godejohann, M.; Spraul, M.; Stuppner, H. Conventional sample enrichment strategies combined with high-performance liquid chromatography-solid phase extraction-nuclear magnetic resonance analysis allows analyte identification from a single minuscule Corydalis solida plant tuber. J. Chromatogr. A 2007, 1163, 138–144. [Google Scholar] [CrossRef]
- Seger, C.; Godejohann, M.; Tseng, L.H.; Spraul, M.; Girtler, A.; Sturm, S.; Stuppner, H. LC-DAD-MS/SPE-NMR hyphenation. A tool for the analysis of pharmaceutically used plant extracts: Identification of isobaric iridoid glycoside regioisomers from Harpagophytum procumbens. Anal. Chem. 2005, 77, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Cogne, A.L.; Queiroz, E.F.; Marston, A.; Wolfender, J.L.; Mavi, S.; Hostettmann, K. On-line identification of unstable iridoids from Jamesbrittenia fodina by HPLC-MS and HPLC-NMR. Phytochem. Anal. 2005, 16, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Roncero, A.M.; Tobal, I.E.; Moro, R.F.; Diez, D.; Marcos, I.S. Halimane diterpenoids: Sources, structures, nomenclature and biological activities. Nat. Prod. Rep. 2018, 35, 955–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.Q.; Yin, Y.P.; Jun, L.; Xuan, L.J. Halimane-type diterpenoids from Vitex rotundifolia and their anti-hyperlipidemia activities. Phytochemistry 2018, 146, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Högner, C.; Sturm, S.; Seger, C.; Stuppner, H. Development and validation of a rapid ultra-high performance liquid chromatography diode array detector method for Vitex agnus-castus. J. Chromatogr. B 2013, 927, 181–190. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Retention Time [min] | [M + Na]+ Experimental [m/z] | [M + Na]+ Accuracy [ppm] | [M + K]+ Experimental [m/z] | [M + K]+ Accuracy [ppm] | Elemental Composition | Mass Shift to VD [u] | Tentative Type of Metabolism a | Substituents by NMR b |
|---|---|---|---|---|---|---|---|---|---|
| M1 | 11.1 | 479.2574 | +2.4 | 495.2319 | −1.7 | C24H40O8 | +50 | 1 * OH, 2 * OH + H | n.i. |
| M2 | 11.6 | 479.2581 | −2.0 | 495.2336 | +3.8 | C24H40O8 | +50 | 1 * OH, 2 * OH + H | OH @ 3β,14,15 |
| M3 | 12.2 | 479.2575 | −0.9 | 495.2337 | +3.6 | C24H40O8 | +50 | 1 * OH, 2 * OH + H | n.i. |
| M4 | 12.4 | 477.2446 | +2.7 | 493.2189 | +1.9 | C24H38O8 | +48 | 3 * OH or 2 * OH and 1 * Ep or 1 * = O and 2 * OH + H | n.i. |
| M5 | 12.9 | 479.2572 | −0.1 | 495.2316 | −1.0 | C24H40O8 | +50 | 1 * OH, 2 * OH + H | OH @ 2β,14,15 |
| M6 | 13.2 | 479.2575 | −0.7 | 495.2321 | −2.0 | C24H40O8 | +50 | 1 * OH, 2 * OH + H | n.i. |
| M7 | 14.3 | 477.2465 | −1.3 | 493.2161 | −1.4 | C24H38O8 | +48 | 3 * OH or 2 * OH and 1* Ep or 1 * = O and 2* OH + H | n.i. |
| M8 | 14.5 | 461.2456 | +2.1 | 477.2246 | −8.5 | C24H38O7 | +32 | 2 * OH or 1 * OH and 1 * Ep | n.i. |
| M9 | 14.7 | 477.2360 | −6.2 | 493.2156 | −0.3 | C24H38O8 | +48 | 3 * OH or 2 * OH and 1 * Ep or 1 * = O and 2 * OH + H | n.i. |
| M10 | 15.0 | 359.2121 | +7.4 | 375.2006 | −0.3 | C20H32O4 | −70 | 2 * deAc and 1 * = O, | n.i. |
| M11 | 15.1 | 361.2280 | −6.2 | 377.2057 | −3.4 | C20H34O4 | −68 | 2 * deAc and 1 * OH or 1 * Ep | n.i. |
| M12 | 15.5 | 461.2453 | +2.8 | 477.2204 | +0.2 | C24H38O7 | +32 | 2 * OH or 1 * OH and 1 * Ep | n.i. |
| M13 | 15.7 | 461.2455 | +2.2 | 477.2214 | −1.8 | C24H38O7 | +32 | 2 * OH or 1 * OH and 1 * Ep | n.i. |
| M14 | 16.5 | 361.2345 | +1.2 | 377.2075 | +3.6 | C20H34O4 | −68 | 2 * deAc and 1 * OH or 1 * Ep | n.i. |
| M15 | 17.1 | 345.2359 | +3.7 | 361.2129 | +2.9 | C20H34O3 | −84 | 2 * deAc | deAc @ 6,7 |
| M16 | 18.1 | 445.2522 | −1.2 | 461.2263 | −1.5 | C24H38O6 | +16 | 1 * OH or 1 * Ep | OH @ 2β |
| M17 | 18.4 | 445.2543 | +4.0 | 461.2285 | +3.2 | C24H38O6 | +16 | 1 * OH or 1 * Ep | OH @ 3β |
| M18 | 18.7 | 463.2652 | +3.1 | 479.2388 | +3.7 | C24H40O7 | +34 | 2 * OH + H | n.i. |
| M19 | 19.0 | 463.2648 | +3.9 | 479.2391 | +3.1 | C24H40O7 | +34 | 2 * OH + H | OH @ 14,15 |
| M20 | 19.6 | 445.2573 | −2.8 | 461.2306 | −1.3 | C24H38O6 | +16 | 1 * OH or 1 * Ep | OH @ 3α |
| M21 | 20.3 | 461.2454 | +2.6 | 477.2211 | −1.3 | C24H38O7 | +32 | 2 * OH or 1 * OH and 1 * Ep | n.i. |
| M22 | 22.0 | 443.2394 | +2.3 | 459.2132 | +2.5 | C24H36O6 | +14 | 1 * = O | = O @ 3 |
| SM1 | 11.6 | 503.2591 | +4.8 | 519.2348 | +1.4 | C26H40O8 | +74 | 2 * deAc, 1 * deHy 1 * GlcA | n.i. |
| SM2 | 11.9 | 503.2609 | +0.6 | 519.2356 | +4.2 | C26H40O8 | +74 | 2 * deAc, 1 * deHy 1 * GlcA | n.i. |
| SM3 | 12.7 | 503.2608 | +0.8 | 519.2346 | +0.8 | C26H40O8 | +74 | 2 * deAc, 1 * deHy 1 * GlcA | n.i. |
| SM4 | 13.6 | 605.2946 | +2.2 | 621.2672 | +0.1 | C30H46O11 | +176 | 1 * GlcA | n.i. |
| SM5 | 20.9 | 387.2489 | +4.4 | 403.2225 | +4.9 | C22H36O4 | −42 | 1 * deAc | n.i. |
| VD | 28.8 | 429.2597 | +3.4 | 445.2339 | +2.7 | C24H38O5 | 0 | parent compound | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sturm, S.; Högner, C.; Seger, C.; Stuppner, H. Combining HPLC-DAD-QTOF-MS and HPLC-SPE-NMR to Monitor In Vitro Vitetrifolin D Phase I and II Metabolism. Metabolites 2021, 11, 529. https://doi.org/10.3390/metabo11080529
Sturm S, Högner C, Seger C, Stuppner H. Combining HPLC-DAD-QTOF-MS and HPLC-SPE-NMR to Monitor In Vitro Vitetrifolin D Phase I and II Metabolism. Metabolites. 2021; 11(8):529. https://doi.org/10.3390/metabo11080529
Chicago/Turabian StyleSturm, Sonja, Christina Högner, Christoph Seger, and Hermann Stuppner. 2021. "Combining HPLC-DAD-QTOF-MS and HPLC-SPE-NMR to Monitor In Vitro Vitetrifolin D Phase I and II Metabolism" Metabolites 11, no. 8: 529. https://doi.org/10.3390/metabo11080529
APA StyleSturm, S., Högner, C., Seger, C., & Stuppner, H. (2021). Combining HPLC-DAD-QTOF-MS and HPLC-SPE-NMR to Monitor In Vitro Vitetrifolin D Phase I and II Metabolism. Metabolites, 11(8), 529. https://doi.org/10.3390/metabo11080529