
TCF19 Impacts a Network of Inflammatory and DNA Damage Response Genes in the Pancreatic β-Cell


Abstract
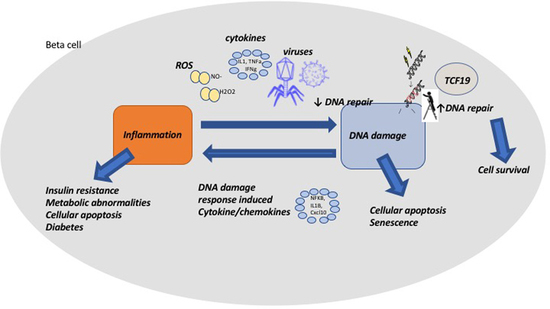
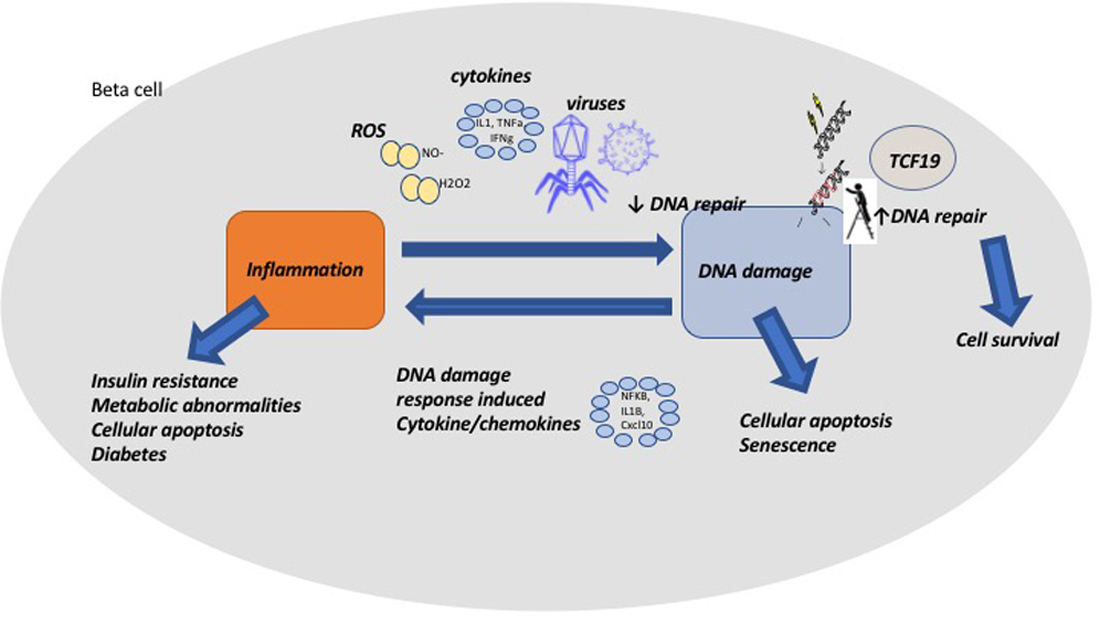{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
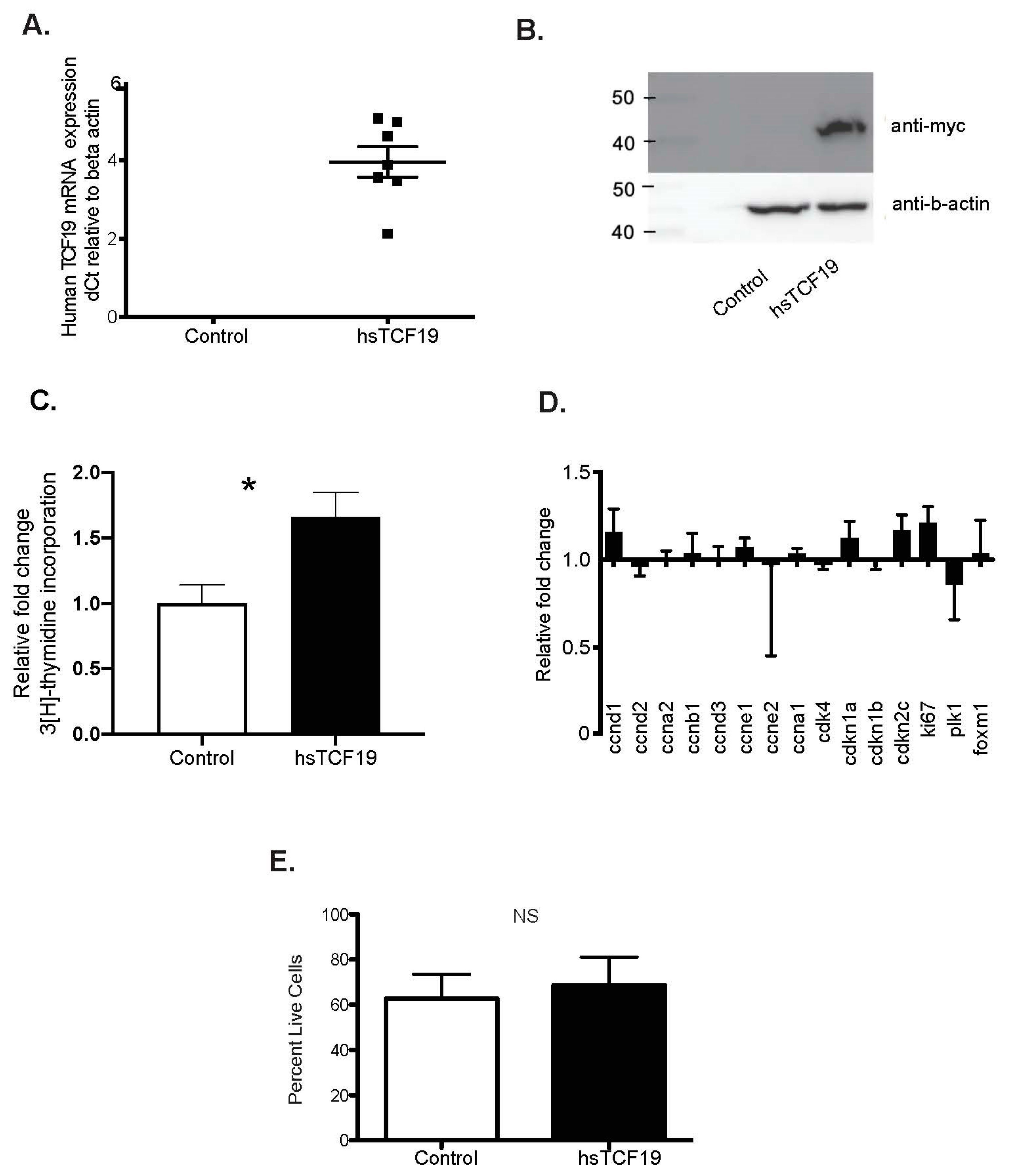
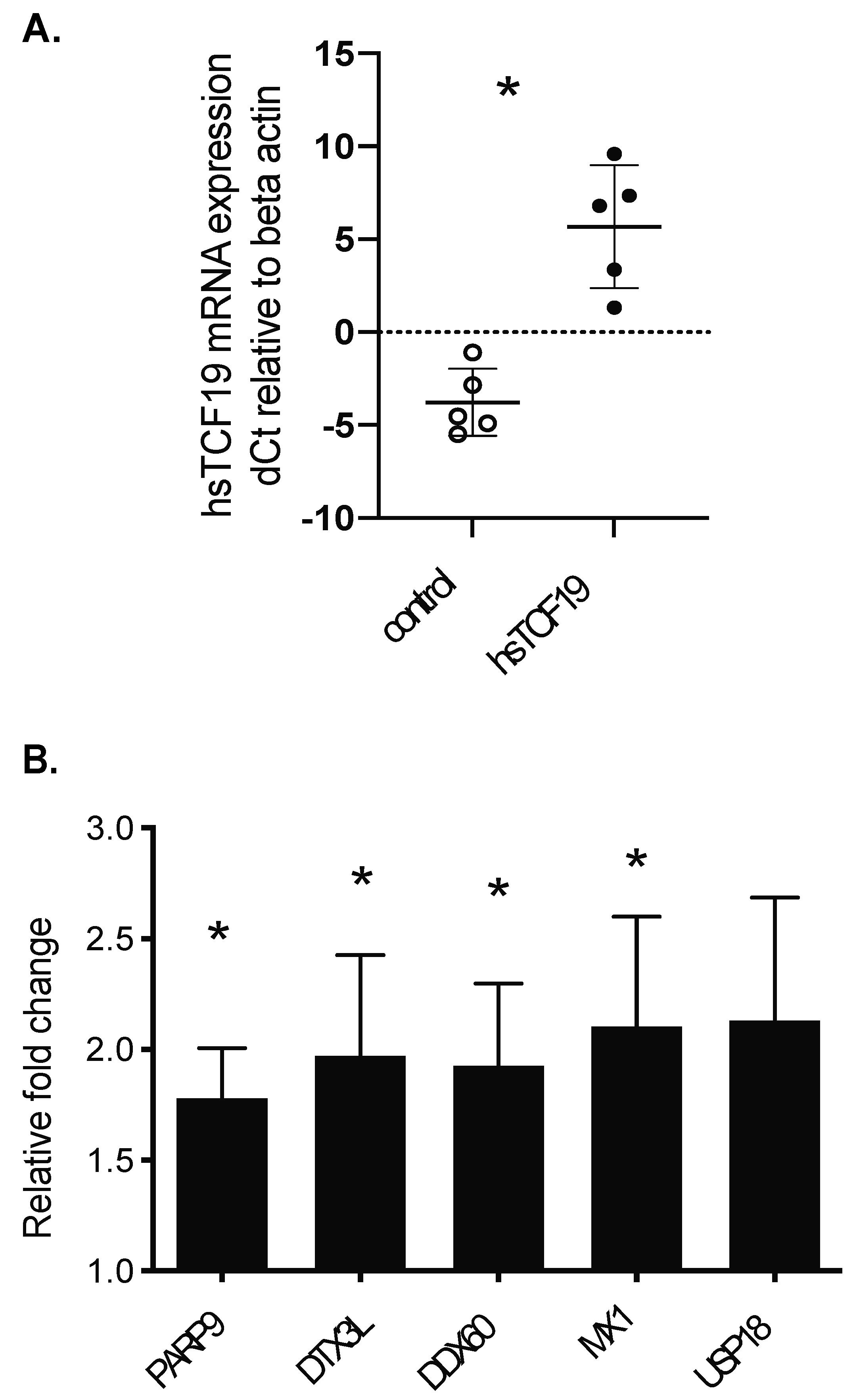
2.1. Human TCF19 Overexpression Increases 3H-Thymidine Incorporation in INS-1 Cells but Does Not Change Cell Cycle Gene Expression
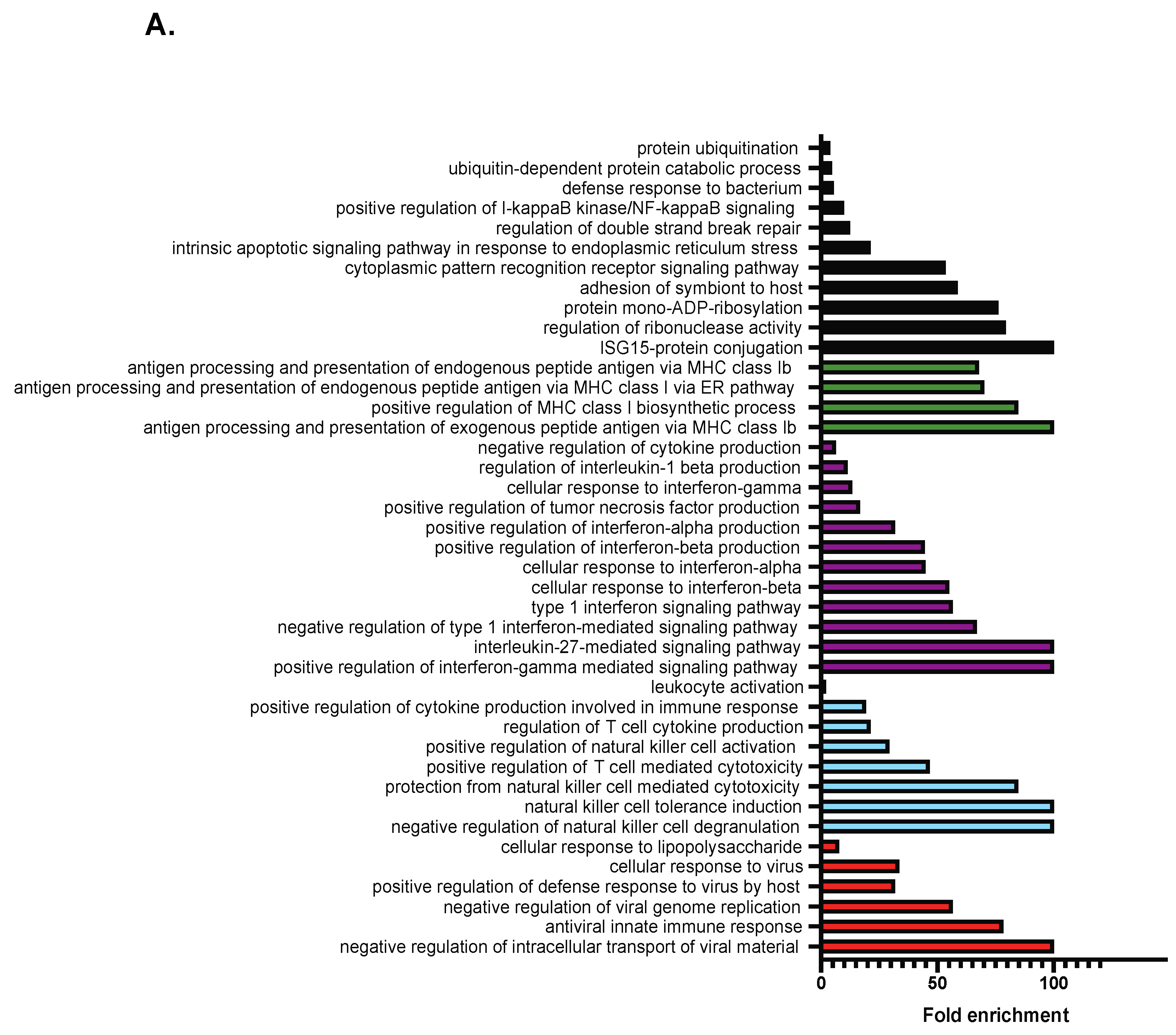
2.2. RNA-Seq Analysis Reveals a Role for TCF19 in Regulating Viral, Inflammatory, and DNA Damage Genes
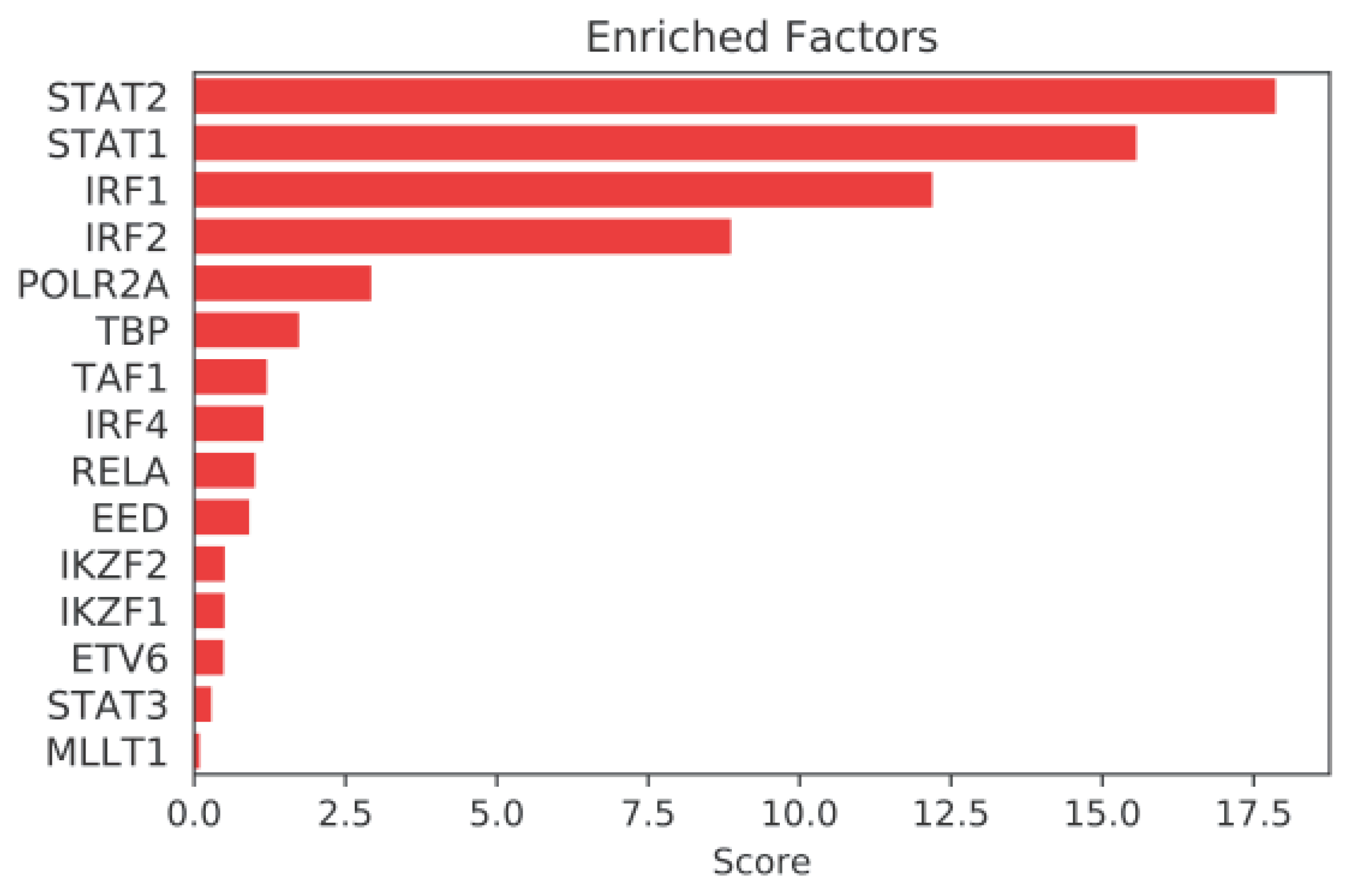
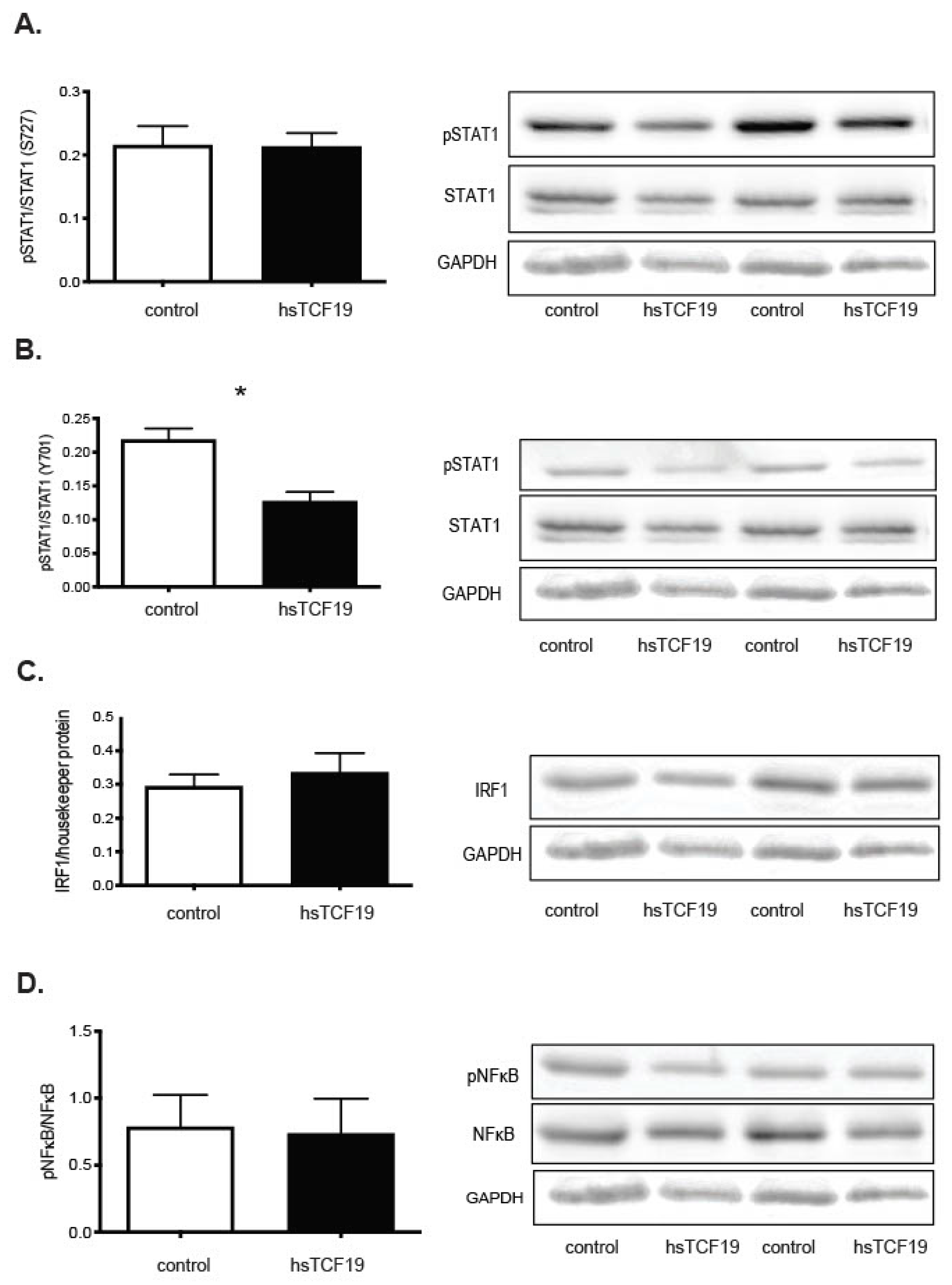
2.3. Mining Algorithm for GenetIc Controllers (MAGIC) Analysis for Common Transcriptional Regulators
3. Discussion
4. Materials and Methods
4.1. Human Islets and INS-1 Cell Culture
4.2. Creation of TCF19 Overexpression Vector
4.3. Transfection with hsTCF19-His/Myc-pcDNA4
4.4. Western Blotting
4.5. Quantitative Real-Time PCR
4.6. Viability
4.7. Proliferation/3H-Thymidine Incorporation
4.8. RNA Sequencing
4.9. GO Term Enrichment
4.10. Protein-Protein Interaction Network Construction
4.11. Mining Algorithm for GenetIc Controllers (MAGIC) Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hinokio, Y.; Suzuki, S.; Hirai, M.; Chiba, M.; Hirai, A.; Toyota, T. Oxidative DNA Damage in Diabetes Mellitus: Its Association with Diabetic Complications. Diabetologia 1999, 42, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, J.; Chen, Y.; Wang, J.J.; Ratan, R.; Zhang, S.X. Activation of Endoplasmic Reticulum Stress by Hyperglycemia Is Essential for Müller Cell–Derived Inflammatory Cytokine Production in Diabetes. Diabetes 2012, 6, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Djuric, Z.; Lu, M.H.; Lewis, S.M.; Luongo, D.A.; Chen, X.W.; Heilbrun, L.K.; Reading, B.A.; Duffy, P.H.; Hart, R.W. Oxidative DNA Damage Levels in Rats Fed Low-Fat, High-Fat, or Calorie-Restricted Diets. Toxicol. Appl. Pharm. 1992, 115, 156–160. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose Toxicity in β-Cells: Type 2 Diabetes, Good Radicals Gone Bad, and the Glutathione Connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic Reticulum Stress, Obesity and Diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.-C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of Pancreatic β-Cell Death in Type 1 and Type 2 Diabetes Many Differences, Few Similarities. Diabetes 2005, 54, S97–S107. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, É.; Sauter, N.S.; Böni-Schnetzler, M. Inflammation in Obesity and Diabetes: Islet Dysfunction and Therapeutic Opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Ron, D. Endoplasmic Reticulum Stress and the Development of Diabetes: A Review. Diabetes 2002, 51, S455–S461. [Google Scholar] [CrossRef]
- Butcher, M.J.; Hallinger, D.; Garcia, E.; Machida, Y.; Chakrabarti, S.; Nadler, J.; Galkina, E.V.; Imai, Y. Association of Proinflammatory Cytokines and Islet Resident Leucocytes with Islet Dysfunction in Type 2 Diabetes. Diabetologia 2014, 57, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.T.; Navidi, A.A.; Rezaei, M.; Babahmadi-Rezaei, H. Oxidative Damage to DNA and Lipids: Correlation with Protein Glycation in Patients with Type 1 Diabetes. J. Clin. Lab. Anal. 2010, 24, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kawamori, D.; Matsuoka, T.; Kajimoto, Y.; Yamasaki, Y. Oxidative Stress and Pancreatic β-Cell Dysfunction. Am. J. Ther. 2005, 12, 529–533. [Google Scholar] [CrossRef]
- Blasiak, J.; Arabski, M.; Krupa, R.; Wozniak, K.; Zadrozny, M.; Kasznicki, J.; Zurawska, M.; Drzewoski, J. DNA Damage and Repair in Type 2 Diabetes Mellitus. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2004, 554, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.; Krogvold, L.; Zhitomirsky, S.; Swisa, A.; Fischman, M.; Lax, T.; Dahan, T.; Hurvitz, N.; Weinberg-Corem, N.; Klochendler, A.; et al. Beta Cell DNA Damage Response Promotes Islet Inflammation in Type 1 Diabetes. Diabetes 2018, 67, 2305–2318. [Google Scholar] [CrossRef]
- Oleson, B.J.; Broniowska, K.A.; Schreiber, K.H.; Tarakanova, V.L.; Corbett, J.A. Nitric Oxide Induces Ataxia Telangiectasia Mutated (ATM) Protein-Dependent ΓH2AX Protein Formation in Pancreatic β Cells. J. Biol. Chem. 2014, 289, 11454–11464. [Google Scholar] [CrossRef] [PubMed]
- Oleson, B.J.; Broniowska, K.A.; Naatz, A.; Hogg, N.; Tarakanova, V.L.; Corbett, J.A. Nitric Oxide Suppresses β-Cell Apoptosis by Inhibiting the DNA Damage Response. Mol. Cell Biol. 2016, 36, 2067–2077. [Google Scholar] [CrossRef]
- Cheung, Y.H.; Watkinson, J.; Anastassiou, D. Conditional Meta-Analysis Stratifying on Detailed HLA Genotypes Identifies a Novel Type 1 Diabetes Locus around TCF19 in the MHC. Hum. Genet. 2011, 129, 161–176. [Google Scholar] [CrossRef]
- Harder, M.N.; Appel, E.V.R.; Grarup, N.; Gjesing, A.P.; Ahluwalia, T.S.; Jørgensen, T.; Christensen, C.; Brandslund, I.; Linneberg, A.; Sørensen, T.I.A.; et al. The Type 2 Diabetes Risk Allele of TMEM154-Rs6813195 Associates with Decreased Beta Cell Function in a Study of 6,486 Danes. PLoS ONE 2015, 10, e0120890. [Google Scholar] [CrossRef]
- Nair, A.K.; Baier, L.J. Complex Genetics of Type 2 Diabetes and Effect Size: What Have We Learned from Isolated Populations? Rev. Diabet. Stud. 2015, 12, 299–319. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Go, M.J.; Zhang, W.; Below, J.E.; Gaulton, K.J.; Ferreira, T.; Horikoshi, M.; Johnson, A.D.; Ng, M.C.; Prokopenko, I.; et al. Genome-Wide Trans-Ancestry Meta-Analysis Provides Insight into the Genetic Architecture of Type 2 Diabetes Susceptibility. Nat. Genet. 2014, 46, 234–244. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Linnemann, A.K.; Fontaine, D.A.; Whillock, A.L.; Harris, T.W.; Schleis, G.J.; Truchan, N.A.; Marty-Santos, L.; Lavine, J.A.; Cleaver, O.; et al. Tcf19 Is a Novel Islet Factor Necessary for Proliferation and Survival in the INS-1 β-Cell Line. Am. J. Physiol. Endoc. 2013, 305, E600–E610. [Google Scholar] [CrossRef]
- Yang, Y.; Gurung, B.; Wu, T.; Wang, H.; Stoffers, D.A.; Hua, X. Reversal of Preexisting Hyperglycemia in Diabetic Mice by Acute Deletion of the Men1 Gene. Proc. Natl. Acad. Sci. USA 2010, 107, 20358–20363. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.P.; Paul, P.K.; Rabaglia, M.E.; Stapleton, D.S.; Schueler, K.L.; Broman, A.T.; Ye, S.I.; Leng, N.; Brandon, C.J.; Neto, E.C.; et al. The Transcription Factor Nfatc2 Regulates β-Cell Proliferation and Genes Associated with Type 2 Diabetes in Mouse and Human Islets. Plos Genet. 2016, 12, e1006466. [Google Scholar] [CrossRef]
- Klochendler, A.; Caspi, I.; Corem, N.; Moran, M.; Friedlich, O.; Elgavish, S.; Nevo, Y.; Helman, A.; Glaser, B.; Eden, A.; et al. The Genetic Program of Pancreatic β-Cell Replication In Vivo. Diabetes 2016, 65, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Sampaio, H.C.; Liu, B.; Drynda, R.; Ledesma, A.M.R.D.; King, A.J.; Bowe, J.E.; Malicet, C.; Iovanna, J.L.; Jones, P.M.; Persaud, S.J.; et al. Nupr1 Deletion Protects against Glucose Intolerance by Increasing Beta Cell Mass. Diabetologia 2013, 56, 2477–2486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ndiaye, F.K.; Ortalli, A.; Canouil, M.; Huyvaert, M.; Salazar-Cardozo, C.; Lecoeur, C.; Verbanck, M.; Pawlowski, V.; Boutry, R.; Durand, E.; et al. Expression and Functional Assessment of Candidate Type 2 Diabetes Susceptibility Genes Identify Four New Genes Contributing to Human Insulin Secretion. Mol. Metab. 2017, 6, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, D.H.; Yaffe, M.B. 14-3-3 Proteins, FHA Domains and BRCT Domains in the DNA Damage Response. DNA Repair 2009, 8, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.A.; Musselman, C.A. Chromatin Signaling and Diseases; Academic Press Inc.: Cambridge, MA, USA, 2016; pp. 127–147. [Google Scholar]
- Sen, S.; Sanyal, S.; Srivastava, D.K.; Dasgupta, D.; Roy, S.; Das, C. Transcription Factor 19 Interacts with Histone 3 Lysine 4 Trimethylation and Controls Gluconeogenesis via the Nucleosome-Remodeling-Deacetylase Complex. J. Biol. Chem. 2017, 292, 20362–20378. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.; Sen, S.; Klein, B.J.; Tiwary, N.; Gadad, S.S.; Kutateladze, T.G.; Roy, S.; Das, C. TCF19 Promotes Cell Proliferation through Binding to the Histone H3K4me3 Mark. Biochemistry 2019, 59, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Limsirichaikul, S.; Niimi, A.; Fawcett, H.; Lehmann, A.; Yamashita, S.; Ogi, T. A Rapid Non-Radioactive Technique for Measurement of Repair Synthesis in Primary Human Fibroblasts by Incorporation of Ethynyl Deoxyuridine (EdU). Nucleic Acids Res. 2009, 37, e31. [Google Scholar] [CrossRef]
- Dasika, G.K.; Lin, S.-C.J.; Zhao, S.; Sung, P.; Tomkinson, A.; Lee, E.Y.-H.P. DNA Damage-Induced Cell Cycle Checkpoints and DNA Strand Break Repair in Development and Tumorigenesis. Oncogene 1999, 18, 7883–7899. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive API. Nucleic Acids Res. 2020, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Thomas, P. Protein Networks and Pathway Analysis. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukar Gene 2014, 24, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Jividen, K.; Spencer, A.; Dworak, N.; Ni, L.; Oostdyk, L.T.; Chatterjee, M.; Kuśmider, B.; Reon, B.; Parlak, M.; et al. Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9. Mol. Cell 2017, 66, 503–516 e5. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Hartmann, R.; Ablasser, A.; Hopfner, K.-P. OAS Proteins and CGAS: Unifying Concepts in Sensing and Responding to Cytosolic Nucleic Acids. Nat. Rev. Immunol. 2014, 14, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Kondratova, A.A.; Cheon, H.; Dong, B.; Holvey-Bates, E.G.; Hasipek, M.; Taran, I.; Gaughan, C.; Jha, B.K.; Silverman, R.H.; Stark, G.R. Suppressing PARylation by 2′,5′-oligoadenylate Synthetase 1 Inhibits DNA Damage-induced Cell Death. Embo. J. 2020, 39, e101573. [Google Scholar] [CrossRef]
- Verhelst, J.; Parthoens, E.; Schepens, B.; Fiers, W.; Saelens, X. Interferon-Inducible Protein Mx1 Inhibits Influenza Virus by Interfering with Functional Viral Ribonucleoprotein Complex Assembly. J. Virol. 2012, 86, 13445–13455. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. DDX60, a DEXD/H Box Helicase, Is a Novel Antiviral Factor Promoting RIG-I-like Receptor-Mediated Signaling. Mol. Cell Biol. 2011, 31, 3802–3819. [Google Scholar] [CrossRef]
- Honke, N.; Shaabani, N.; Zhang, D.-E.; Hardt, C.; Lang, K.S. Multiple Functions of USP18. Cell Death Dis. 2016, 7, e2444. [Google Scholar] [CrossRef] [PubMed]
- Roopra, A. MAGIC: A Tool for Predicting Transcription Factors and Cofactors Driving Gene Sets Using ENCODE Data. PLoS Comput. Biol. 2020, 16, e1007800. [Google Scholar] [CrossRef]
- Sadzak, I.; Schiff, M.; Gattermeier, I.; Glinitzer, R.; Sauer, I.; Saalmüller, A.; Yang, E.; Schaljo, B.; Kovarik, P. Recruitment of Stat1 to Chromatin Is Required for Interferon-Induced Serine Phosphorylation of Stat1 Transactivation Domain. Proc. Natl. Acad. Sci. USA 2008, 105, 8944–8949. [Google Scholar] [CrossRef] [PubMed]
- Varinou, L.; Ramsauer, K.; Karaghiosoff, M.; Kolbe, T.; Pfeffer, K.; Müller, M.; Decker, T. Phosphorylation of the Stat1 Transactivation Domain Is Required for Full-Fledged IFN-γ-Dependent Innate Immunity. Immunity 2003, 19, 793–802. [Google Scholar] [CrossRef]
- Cai, X.; Chiu, Y.-H.; Chen, Z.J. The CGAS-CGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Analysis of Islet Inflammation in Human Type 1 Diabetes. Clin. Exp. Immunol. 2009, 155, 173–181. [Google Scholar] [CrossRef]
- Åkerblom, H.K.; Vaarala, O.; Hyöty, H.; Ilonen, J.; Knip, M. Environmental Factors in the Etiology of Type 1 Diabetes. Am. J. Med. Genet. 2002, 115, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Becattini, B. JNK at the Crossroad of Obesity, Insulin Resistance, and Cell Stress Response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Tatsch, E.; Carvalho, J.A.M.D.; Hausen, B.S.; Bollick, Y.S.; Torbitz, V.D.; Duarte, T.; Scolari, R.; Duarte, M.M.M.F.; Londero, S.W.K.; Vaucher, R.A.; et al. Oxidative DNA Damage Is Associated with Inflammatory Response, Insulin Resistance and Microvascular Complications in Type 2 Diabetes. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 782, 17–22. [Google Scholar] [CrossRef]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Tavana, O.; Puebla-Osorio, N.; Sang, M.; Zhu, C. Absence of P53-Dependent Apoptosis Combined With Nonhomologous End-Joining Deficiency Leads to a Severe Diabetic Phenotype in Mice. Diabetes 2010, 59, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Brzostek-Racine, S.; Gordon, C.; Scoy, S.V.; Reich, N.C. The DNA Damage Response Induces IFN. J. Immunol. 2011, 187, 5336–5345. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Dunphy, G. CGAS-Independent STING Activation in Response to DNA Damage. Mol. Cell Oncol. 2019, 6, 1–3. [Google Scholar] [CrossRef]
- Bai, J.; Liu, F. The CGAS-CGAMP-STING Pathway: A Molecular Link between Immunity and Metabolism. Diabetes 2019, 68, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Wållberg, M.; Cooke, A. Immune Mechanisms in Type 1 Diabetes. Trends Immunol. 2013, 34, 583–591. [Google Scholar] [CrossRef]
- Mboko, W.P.; Mounce, B.C.; Wood, B.M.; Kulinski, J.M.; Corbett, J.A.; Tarakanova, V.L. Coordinate Regulation of DNA Damage and Type I Interferon Responses Imposes an Antiviral State That Attenuates Mouse Gammaherpesvirus Type 68 Replication in Primary Macrophages. J. Virol. 2012, 86, 6899–6912. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-Induced DNA Damage, Mutations and Cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef]
- Frontini, M.; Vijayakumar, M.; Garvin, A.; Clarke, N. A ChIP–Chip Approach Reveals a Novel Role for Transcription Factor IRF1 in the DNA Damage Response. Nucleic Acids Res. 2009, 37, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Henriksen, M.A.; Takeuchi, K.; Schaefer, O.; Liu, B.; Hoeve, J.t.; Ren, Z.; Mao, X.; Chen, X.; Shuai, K.; et al. Implications of an Antiparallel Dimeric Structure of Nonphosphorylated STAT1 for the Activation–Inactivation Cycle. Proc. Natl. Acad. Sci. USA. 2005, 102, 3966–3971. [Google Scholar] [CrossRef]
- Durocher, D.; Jackson, S.P. The FHA Domain. Febs. Lett. 2002, 513, 58–66. [Google Scholar] [CrossRef]
- Li, J.; Lee, G.I.; Doren, S.R.V.; Walker, J.C. The FHA Domain Mediates Phosphoprotein Interactions. J. Cell Sci. 2000, 23, 4143–4149. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.H.; Fontaine, D.A.; Lodh, S.; Blumer, J.T.; Roopra, A.; Davis, D.B. TCF19 Impacts a Network of Inflammatory and DNA Damage Response Genes in the Pancreatic β-Cell. Metabolites 2021, 11, 513. https://doi.org/10.3390/metabo11080513
Yang GH, Fontaine DA, Lodh S, Blumer JT, Roopra A, Davis DB. TCF19 Impacts a Network of Inflammatory and DNA Damage Response Genes in the Pancreatic β-Cell. Metabolites. 2021; 11(8):513. https://doi.org/10.3390/metabo11080513
Chicago/Turabian StyleYang, Grace H., Danielle A. Fontaine, Sukanya Lodh, Joseph T. Blumer, Avtar Roopra, and Dawn Belt Davis. 2021. "TCF19 Impacts a Network of Inflammatory and DNA Damage Response Genes in the Pancreatic β-Cell" Metabolites 11, no. 8: 513. https://doi.org/10.3390/metabo11080513
APA StyleYang, G. H., Fontaine, D. A., Lodh, S., Blumer, J. T., Roopra, A., & Davis, D. B. (2021). TCF19 Impacts a Network of Inflammatory and DNA Damage Response Genes in the Pancreatic β-Cell. Metabolites, 11(8), 513. https://doi.org/10.3390/metabo11080513