
Fructose and Mannose in Inborn Errors of Metabolism and Cancer

Abstract
1. Introduction
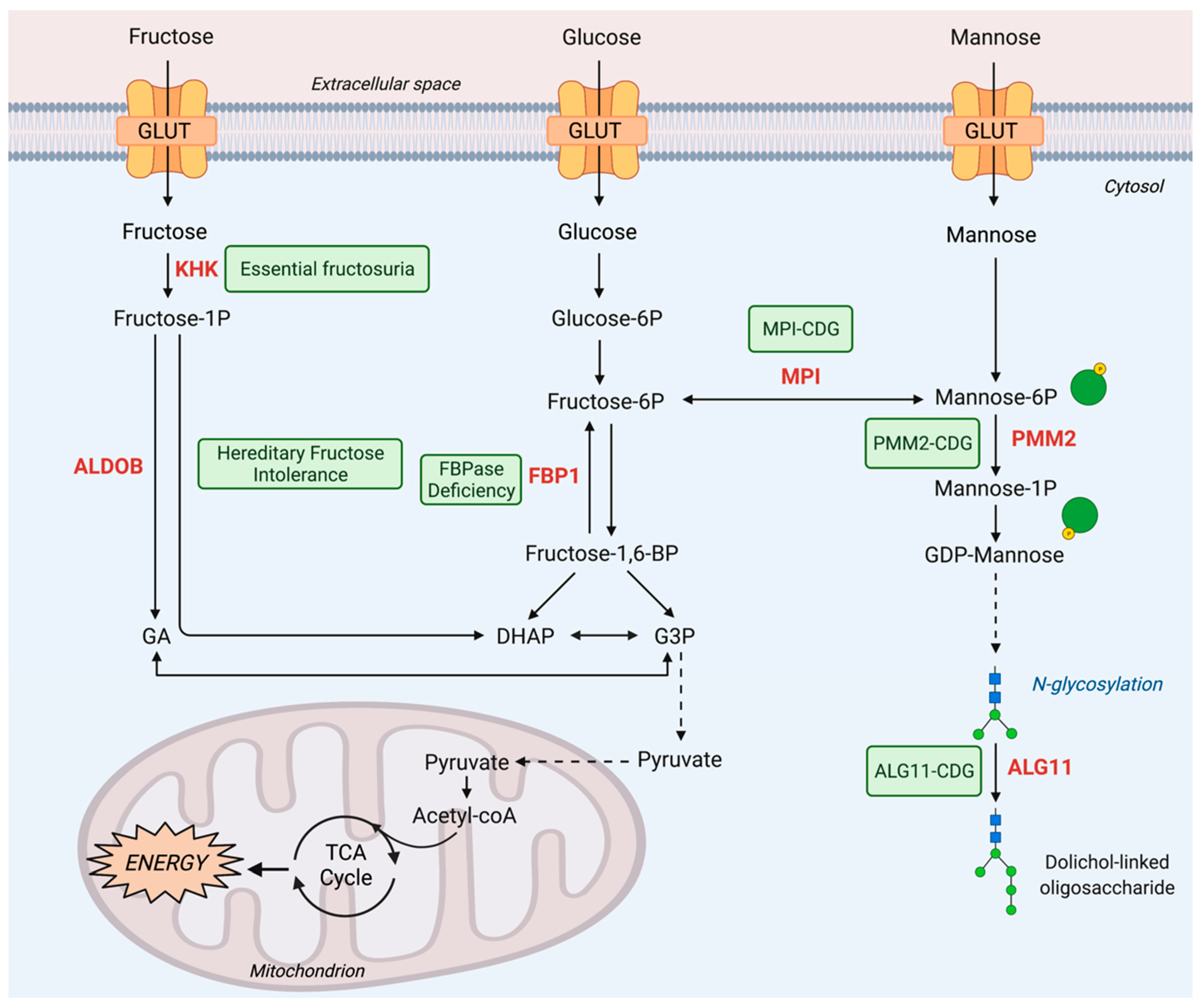
2. Fructose Metabolism Disorders
2.1. Inborn Errors of Fructose Metabolism
2.1.1. Essential Fructosuria
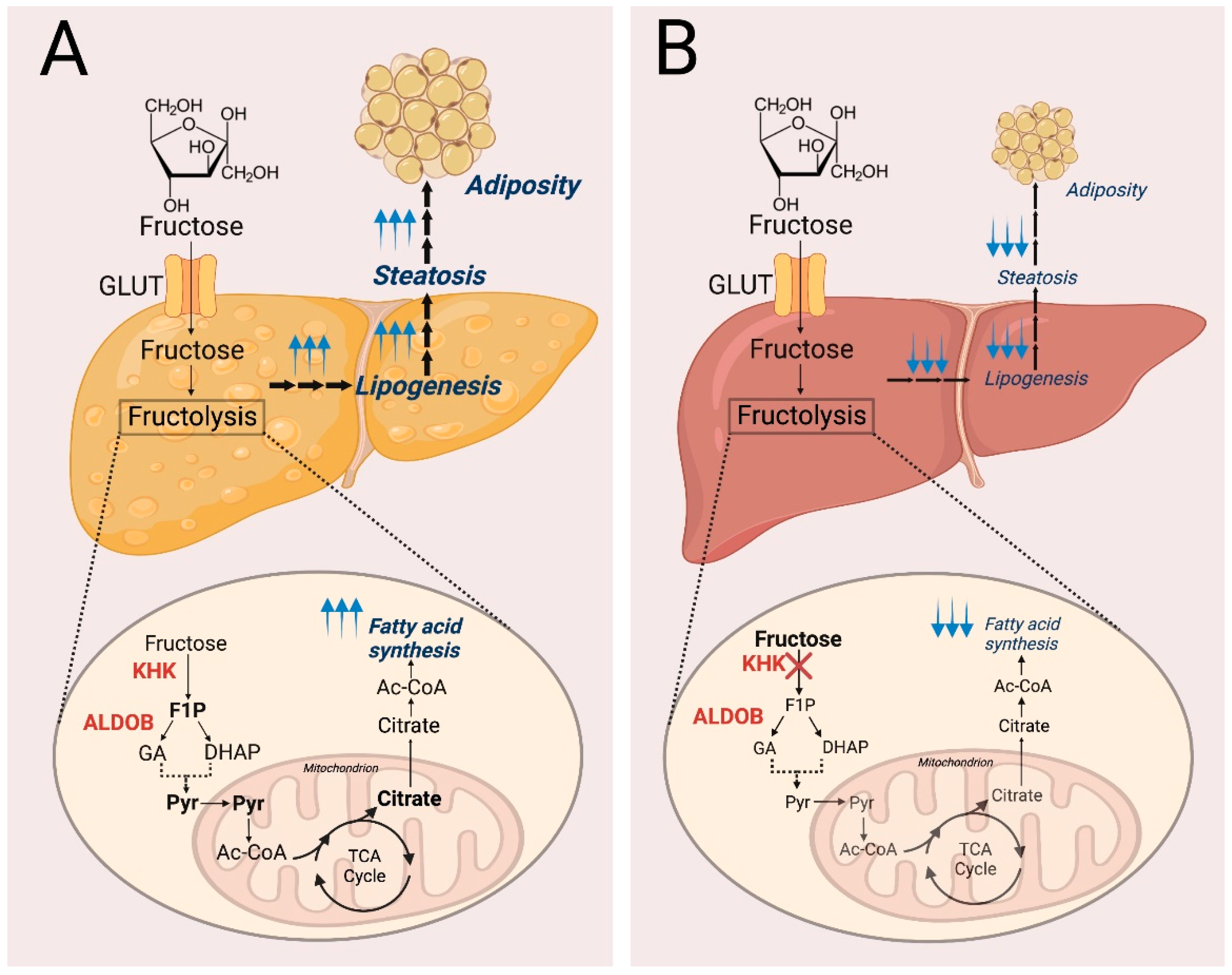
2.1.2. Hereditary Fructose Intolerance
2.1.3. FBPase Deficiency
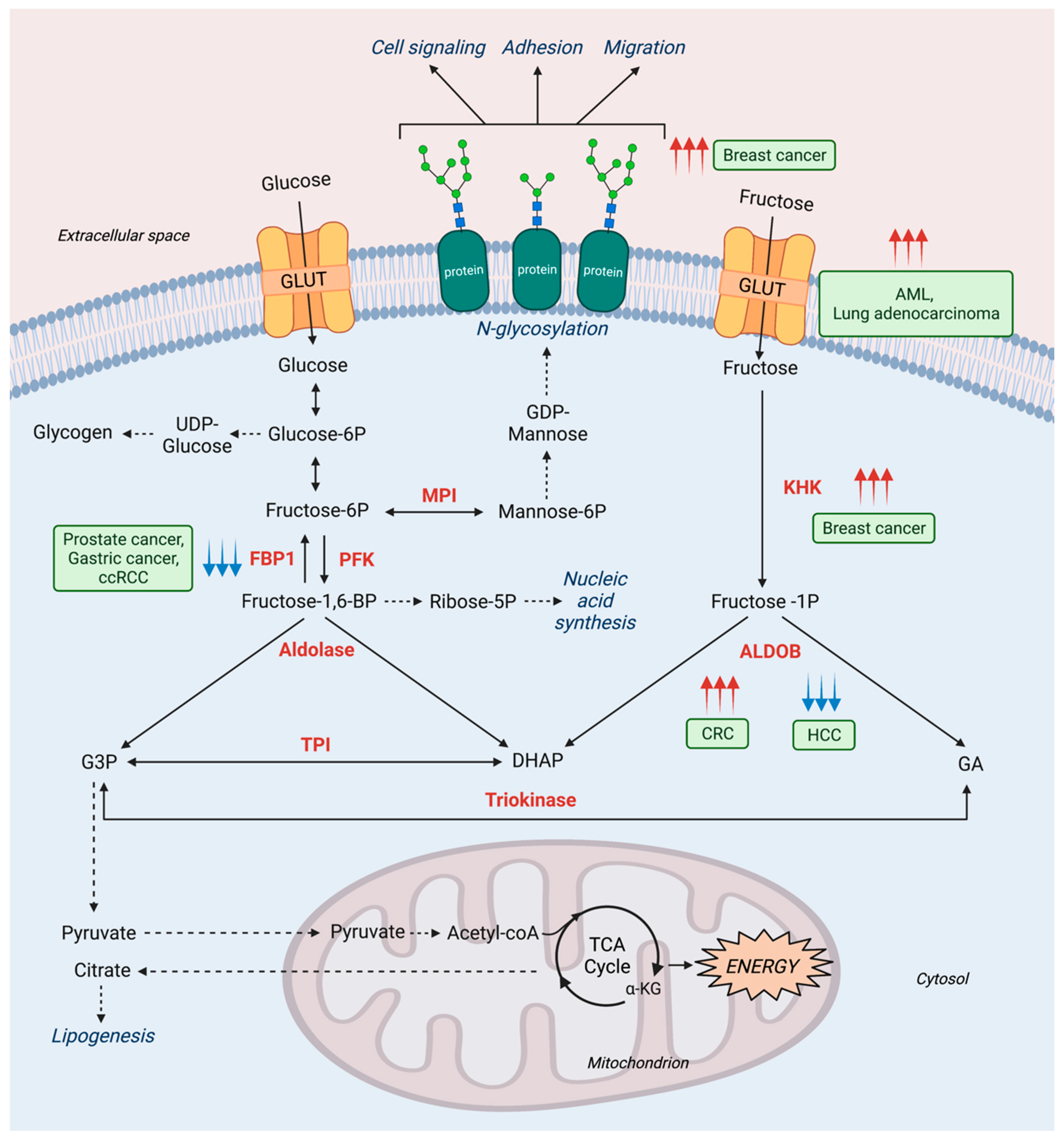
2.2. Fructose Metabolism in Cancer
3. Mannose Metabolism Disorders
3.1. Inborn Errors of Mannose Metabolism
3.1.1. MPI-CDG
3.1.2. PMM2-CDG
3.1.3. ALG11-CDG
3.2. Mannose Metabolism and Other Diseases
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.H.; Elhassan, G.O.; Ibrahim, M.E.; Orozco, D.P.J.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef] [PubMed]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Lu, C.; Tutnauer, J.; Hartman, T.E.; Hwang, S.K.; Murphy, C.J.; Pauli, C.; Morris, R.; Taylor, S.; Bosch, K.; et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019, 363, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Alton, G.; Hasilik, M.; Niehues, R.; Panneerselvam, K.; Etchison, J.R.; Fana, F.; Freeze, H.H. Direct utilization of mannose for mammalian glycoprotein biosynthesis. Glycobiology 1998, 8, 285–295. [Google Scholar] [CrossRef]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—what isotopic tracer studies tell us. Nutr. Metab. 2012, 9, 89. [Google Scholar] [CrossRef]
- Johnson, R.J.; Perez-Pozo, S.E.; Lillo, J.L.; Grases, F.; Schold, J.D.; Kuwabara, M.; Sato, Y.; Hernando, A.A.; Garcia, G.; Jensen, T.; et al. Fructose increases risk for kidney stones: Potential role in metabolic syndrome and heat stress. BMC Nephrol. 2018, 19, 315. [Google Scholar] [CrossRef]
- Hu, X.; Shi, Y.; Zhang, P.; Miao, M.; Zhang, T.; Jiang, B. d-Mannose: Properties, Production, and Applications: An Overview. Compr. Rev. Food Sci. Food Saf. 2016, 15, 773–785. [Google Scholar] [CrossRef]
- Mussatto, S.I.; Carneiro, L.M.; Silva, J.P.A.; Roberto, I.C.; Teixeira, J.A. A study on chemical constituents and sugars extraction from spent coffee grounds. Carbohydr. Polym. 2011, 83, 368–374. [Google Scholar] [CrossRef]
- Somboonkaew, N.; Terry, L.A. Physiological and biochemical profiles of imported litchi fruit under modified atmosphere packaging. Postharvest Biol. Technol. 2010, 56, 246–253. [Google Scholar] [CrossRef]
- Chatterjee, A.K.; Montgomery, R. The carbohydrate of ovomucoid. Arch. Biochem. Biophys. 1962, 99, 426–432. [Google Scholar] [CrossRef]
- Jaeken, J.; Péanne, R. What is new in CDG? J. Inherit. Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef]
- Asipu, A.; Hayward, B.E.; O’Reilly, J.; Bonthron, D.T. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes 2003, 52, 2426–2432. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and, A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e353. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.O.; Yang, X.; Lu, K.; Cao, J.; Herath, K.; Rosahl, T.W.; Askew, R.; Pavlovic, G.; Zhou, G.; Li, C.; et al. Ketohexokinase knockout mice, a model for essential fructosuria, exhibit altered fructose metabolism and are protected from diet-induced metabolic defects. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E386–E393. [Google Scholar] [CrossRef]
- Froesch, E.R. Disorders of fructose metabolism. Clin. Endocrinol. Metab. 1976, 5, 599–611. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Li, N.; Cicerchi, C.; Inaba, S.; Chen, W.; Roncal-Jimenez, C.; Le, M.T.; Wempe, M.F.; Milagres, T.; Ishimoto, T.; et al. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat. Commun. 2017, 8, 14181. [Google Scholar] [CrossRef]
- Laron, Z. Essential benign fructosuria. Arch. Dis. Child 1961, 36, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Bonthron, D.T.; Brady, N.; Donaldson, I.A.; Steinmann, B. Molecular basis of essential fructosuria: Molecular cloning and mutational analysis of human ketohexokinase (fructokinase). Hum. Mol. Genet. 1994, 3, 1627–1631. [Google Scholar] [CrossRef]
- Boesiger, P.; Buchli, R.; Meier, D.; Steinmann, B.; Gitzelmann, R. Changes of liver metabolite concentrations in adults with disorders of fructose metabolism after intravenous fructose by 31P magnetic resonance spectroscopy. Pediatr. Res. 1994, 36, 436–440. [Google Scholar] [CrossRef]
- Oppelt, S.A.; Sennott, E.M.; Tolan, D.R. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Mol. Genet. Metab. 2015, 114, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.; Snodgrass, G.J.; Oberholzer, V.G.; Burgess, E.A.; Dobbs, R.H. Fructosaemia. Observations on seven cases. Am. J. Med. 1968, 45, 826–838. [Google Scholar] [CrossRef]
- Ahmad, U.; Sharma, J. Fructose 1-Phosphate Aldolase Deficiency; StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557761/ (accessed on 1 May 2021).
- Melkonian, E.A.; Asuka, E.; Schury, M.P. Physiology, Gluconeogenesis; StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541119/ (accessed on 1 May 2021).
- Simons, N.; Debray, F.G.; Schaper, N.C.; Kooi, M.E.; Feskens, E.J.M.; Hollak, C.E.M.; Lindeboom, L.; Koek, G.H.; Bons, J.A.P.; Lefeber, D.J.; et al. Patients with Aldolase B. Deficiency Are Characterized by Increased Intrahepatic Triglyceride Content. J. Clin. Endocrinol. Metab. 2019, 104, 5056–5064. [Google Scholar] [CrossRef] [PubMed]
- Aldámiz-Echevarría, L.; de Las Heras, J.; Couce, M.L.; Alcalde, C.; Vitoria, I.; Bueno, M.; Blasco-Alonso, J.; Concepción García, M.; Ruiz, M.; Suárez, R.; et al. Non-alcoholic fatty liver in hereditary fructose intolerance. Clin. Nutr. 2020, 39, 455–459. [Google Scholar] [CrossRef]
- Jaeken, J.; Pirard, M.; Adamowicz, M.; Pronicka, E.; van Schaftingen, E. Inhibition of phosphomannose isomerase by fructose 1-phosphate: An explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr. Res. 1996, 40, 764–766. [Google Scholar] [CrossRef]
- Cross, N.C.; de Franchis, R.; Sebastio, G.; Dazzo, C.; Tolan, D.R.; Gregori, C.; Odievre, M.; Vidailhet, M.; Romano, V.; Mascali, G.; et al. Molecular analysis of aldolase B genes in hereditary fructose intolerance. Lancet 1990, 335, 306–309. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Romano, S.; Mention, K.; Vassault, A.; Rabier, D.; Polak, M.; Robert, J.J.; de Keyzer, Y.; de Lonlay, P. What’s new in metabolic and genetic hypoglycaemias: Diagnosis and management. Eur. J. Pediatr. 2008, 167, 257–265. [Google Scholar] [CrossRef]
- Schrodi, S.J.; DeBarber, A.; He, M.; Ye, Z.; Peissig, P.; Van Wormer, J.J.; Haws, R.; Brilliant, M.H.; Steiner, R.D. Prevalence estimation for monogenic autosomal recessive diseases using population-based genetic data. Hum. Genet. 2015, 134, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Rellos, P.; Cox, T.M. Hereditary fructose intolerance. J. Med. Genet. 1998, 35, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Froesch, E.R.; Wolf, H.P.; Baitsch, H.; Prader, A.; Labhart, A. Hereditary fructose intolerance. An inborn defect of hepatic fructose-1-phosphate splitting aldolase. Am. J. Med. 1963, 34, 151–167. [Google Scholar] [CrossRef]
- Coffee, E.M.; Yerkes, L.; Ewen, E.P.; Zee, T.; Tolan, D.R. Increased prevalence of mutant null alleles that cause hereditary fructose intolerance in the American population. J. Inherit. Metab. Dis. 2010, 33, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Coffee, E.M.; Tolan, D.R. Mutations in the promoter region of the aldolase, B. gene that cause hereditary fructose intolerance. J. Inherit. Metab. Dis. 2010, 33, 715–725. [Google Scholar] [CrossRef]
- Baerlocher, K.; Gitzelmann, R.; Steinmann, B.; Gitzelmann-Cumarasamy, N. Hereditary fructose intolerance in early childhood: A major diagnostic challenge. Survey of 20 symptomatic cases. Helv. Paediatr. Acta 1978, 33, 465–487. [Google Scholar]
- Martin, C.R.; Ling, P.R.; Blackburn, G.L. Review of Infant Feeding: Key Features of Breast Milk and Infant Formula. Nutrients 2016, 8, 279. [Google Scholar] [CrossRef]
- Kim, A.Y.; Hughes, J.J.; Pipitone Dempsey, A.; Sondergaard Schatz, K.; Wang, T.; Gunay-Aygun, M. Pitfalls in the Diagnosis of Hereditary Fructose Intolerance. Pediatrics 2020, 146, e20193324. [Google Scholar] [CrossRef]
- Di Dato, F.; Spadarella, S.; Puoti, M.G.; Caprio, M.G.; Pagliardini, S.; Zuppaldi, C.; Vallone, G.; Fecarotta, S.; Esposito, G.; Iorio, R.; et al. Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance. Nutrients 2019, 11, 2397. [Google Scholar] [CrossRef]
- Van den Berghe, G. Disorders of fructose metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment; Fernandes, J., Saudubray, J.-M., Van den Berghe, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 110–116. [Google Scholar]
- Tran, C. Inborn Errors of Fructose Metabolism. What Can We Learn from Them? Nutrients 2017, 9, 356. [Google Scholar] [CrossRef]
- Bouteldja, N.; Timson, D.J. The biochemical basis of hereditary fructose intolerance. J. Inherit. Metab. Dis. 2010, 33, 105–112. [Google Scholar] [CrossRef]
- Plaisance, E.P.; Greenway, F.L.; Boudreau, A.; Hill, K.L.; Johnson, W.D.; Krajcik, R.A.; Perrone, C.E.; Orentreich, N.; Cefalu, W.T.; Gettys, T.W.; et al. Dietary methionine restriction increases fat oxidation in obese adults with metabolic syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E836–E840. [Google Scholar] [CrossRef] [PubMed]
- Simons, N.; Debray, F.G.; Schaper, N.C.; Feskens, E.J.M.; Hollak, C.E.M.; Bons, J.A.P.; Bierau, J.; Houben, A.; Schalkwijk, C.G.; Stehouwer, C.D.A. Kidney and vascular function in adult patients with hereditary fructose intolerance. Mol. Genet. Metab. Rep. 2020, 23, 100600. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Andres-Hernando, A.; Orlicky, D.J.; Cicerchi, C.; Jang, C.; Li, N.; Milagres, T.; Kuwabara, M.; Wempe, M.F.; Rabinowitz, J.D.; et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Investig. 2018, 128, 2226–2238. [Google Scholar] [CrossRef]
- Lebigot, E.; Brassier, A.; Zater, M.; Imanci, D.; Feillet, F.; Thérond, P.; de Lonlay, P.; Boutron, A. Fructose 1,6-bisphosphatase deficiency: Clinical, biochemical and genetic features in French patients. J. Inherit. Metab. Dis. 2015, 38, 881–887. [Google Scholar] [CrossRef]
- Bijarnia-Mahay, S.; Bhatia, S.; Arora, V. Fructose-1,6-Bisphosphatase Deficiency. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Baker, L.; Winegrad, A.I. Fasting hypoglycaemia and metabolic acidosis associated with deficiency of hepatic fructose-1,6-diphosphatase activity. Lancet 1970, 2, 13–16. [Google Scholar] [CrossRef]
- Moey, L.H.; Abdul Azize, N.A.; Yakob, Y.; Leong, H.Y.; Keng, W.T.; Chen, B.C.; Ngu, L.H. Fructose-1,6-bisphosphatase deficiency as a cause of recurrent hypoglycemia and metabolic acidosis: Clinical and molecular findings in Malaysian patients. Pediatr. Neonatol. 2018, 59, 397–403. [Google Scholar] [CrossRef]
- Steinmann, B.; Santer, R.; van den Berghe, G. Disorders of Fructose Metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment; Fernandes, J., Saudubray, J.-M., van den Berghe, G., Walter, J.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 135–142. [Google Scholar]
- Kamate, M.; Jambagi, M.; Gowda, P.; Sonoli, S. Fructose-1,6-diphosphatase deficiency: A treatable neurometabolic disorder. BMJ Case Rep. 2014, 2014, bcr2013201553. [Google Scholar] [CrossRef]
- Li, N.; Chang, G.; Xu, Y.; Ding, Y.; Li, G.; Yu, T.; Qing, Y.; Li, J.; Shen, Y.; Wang, J.; et al. Clinical and Molecular Characterization of Patients with Fructose 1,6-Bisphosphatase Deficiency. Int. J. Mol. Sci. 2017, 18, 857. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Kim, J.H.; Han, J.H.; Ko, S.H.; Ahn, Y.B.; Kim, J.H.; Yang, S.H.; Song, K.H. Novel compound heterozygous mutations in the fructose-1,6-bisphosphatase gene cause hypoglycemia and lactic acidosis. Metabolism 2011, 60, 107–113. [Google Scholar] [CrossRef]
- Nakai, A.; Shigematsu, Y.; Liu, Y.Y.; Kikawa, Y.; Sudo, M. Urinary sugar phosphates and related organic acids in fructose-1,6-diphosphatase deficiency. J. Inherit. Metab. Dis. 1993, 16, 408–414. [Google Scholar] [CrossRef]
- Bhai, P.; Bijarnia-Mahay, S.; Puri, R.D.; Saxena, R.; Gupta, D.; Kotecha, U.; Sachdev, A.; Gupta, D.; Vyas, V.; Agarwal, D.; et al. Clinical and molecular characterization of Indian patients with fructose-1, 6-bisphosphatase deficiency: Identification of a frequent variant (E281K). Ann. Hum. Genet. 2018, 82, 309–317. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Kikawa, Y.; Miyamaoto, J.; Sugimoto, S.; Adachi, M.; Ohura, T.; Mayumi, M. Intravenous glycerol therapy should not be used in patients with unrecognized fructose-1,6-bisphosphatase deficiency. Pediatr. Int. 2003, 45, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Krause, N.; Wegner, A. Fructose Metabolism in Cancer. Cells 2020, 9, 2635. [Google Scholar] [CrossRef] [PubMed]
- Port, A.M.; Ruth, M.R.; Istfan, N.W. Fructose consumption and cancer: Is there a connection? Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Strober, J.W.; Brady, M.J. Dietary Fructose Consumption and Triple-Negative Breast Cancer Incidence. Front. Endocrinol. 2019, 10, 367. [Google Scholar] [CrossRef]
- Aune, D.; Chan, D.S.M.; Vieira, A.R.; Navarro Rosenblatt, D.A.; Vieira, R.; Greenwood, D.C.; Cade, J.E.; Burley, V.J.; Norat, T. Dietary fructose, carbohydrates, glycemic indices and pancreatic cancer risk: A systematic review and meta-analysis of cohort studies. Ann. Oncol. 2012, 23, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Wang, Y.Y.; Zhao, A.; Xia, L.; Xie, G.; Su, M.; Zhao, L.; Liu, J.; Qu, C.; Wei, R.; et al. Enhanced Fructose Utilization Mediated by SLC2A5 Is a Unique Metabolic Feature of Acute Myeloid Leukemia with Therapeutic Potential. Cancer Cell 2016, 30, 779–791. [Google Scholar] [CrossRef]
- Weng, Y.; Fan, X.; Bai, Y.; Wang, S.; Huang, H.; Yang, H.; Zhu, J.; Zhang, F. SLC2A5 promotes lung adenocarcinoma cell growth and metastasis by enhancing fructose utilization. Cell Death Discov. 2018, 4, 38. [Google Scholar] [CrossRef]
- Monzavi-Karbassi, B.; Hine, R.J.; Stanley, J.S.; Ramani, V.P.; Carcel-Trullols, J.; Whitehead, T.L.; Kelly, T.; Siegel, E.R.; Artaud, C.; Shaaf, S.; et al. Fructose as a carbon source induces an aggressive phenotype in MDA-MB-468 breast tumor cells. Int. J. Oncol. 2010, 37, 615–622. [Google Scholar] [CrossRef][Green Version]
- Dennis, J.W.; Granovsky, M.; Warren, C.E. Protein glycosylation in development and disease. Bioessays 1999, 21, 412–421. [Google Scholar] [CrossRef]
- Jeong, S.; Savino, A.M.; Chirayil, R.; Barin, E.; Cheng, Y.; Park, S.M.; Schurer, A.; Mullarky, E.; Cantley, L.C.; Kharas, M.G.; et al. High Fructose Drives the Serine Synthesis Pathway in Acute Myeloid Leukemic Cells. Cell Metab. 2021, 33, 145–159.e146. [Google Scholar] [CrossRef]
- Kim, J.; Kang, J.; Kang, Y.L.; Woo, J.; Kim, Y.; Huh, J.; Park, J.W. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun. 2020, 11, 5436. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.e1244. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.F.; Yuan, S.X.; Yang, F.; Yang, S.; Yang, Y.; Yuan, J.H.; Wang, Z.G.; Xu, Q.G.; Lin, K.Y.; Cai, J.; et al. Aldolase B. inhibits metastasis through Ten-Eleven Translocation 1 and serves as a prognostic biomarker in hepatocellular carcinoma. Mol. Cancer 2015, 14, 170. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Liu, K.L.; Yang, Z.; Lu, B.S.; Qi, J.C.; Han, Z.W.; Yin, Y.W.; Zhang, M.; Chen, D.M.; Wang, X.W.; et al. The involvement of FBP1 in prostate cancer cell epithelial mesenchymal transition, invasion and metastasis by regulating the MAPK signaling pathway. Cell Cycle 2019, 18, 2432–2446. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, J.; Chen, Y.; Cao, W.; Lu, Y.; Yang, J.; Xing, E. Snail Enhances Glycolysis in the Epithelial-Mesenchymal Transition Process by Targeting FBP1 in Gastric Cancer. Cell Physiol. Biochem. 2017, 43, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; van Eijk, H.G.; van der Heul, C.; Corbeel, L.; Eeckels, R.; Eggermont, E. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin. Chim. Acta 1984, 144, 245–247. [Google Scholar] [CrossRef]
- Jaeken, J.; Hennet, T.; Matthijs, G.; Freeze, H.H. CDG nomenclature: Time for a change! Biochim. Biophys. Acta 2009, 1792, 825–826. [Google Scholar] [CrossRef]
- Jaeken, J.; Matthijs, G. Congenital disorders of glycosylation. Annu. Rev. Genom. Hum. Genet. 2001, 2, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H.; Aebi, M. Altered glycan structures: The molecular basis of congenital disorders of glycosylation. Curr. Opin. Struct. Biol. 2005, 15, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Ichikawa, M.; Freeze, H.H. Mannose metabolism: More than meets the eye. Biochem. Biophys. Res. Commun. 2014, 453, 220–228. [Google Scholar] [CrossRef]
- Panneerselvam, K.; Freeze, H.H. Mannose enters mammalian cells using a specific transporter that is insensitive to glucose. J. Biol. Chem. 1996, 271, 9417–9421. [Google Scholar] [CrossRef]
- Niehues, R.; Hasilik, M.; Alton, G.; Körner, C.; Schiebe-Sukumar, M.; Koch, H.G.; Zimmer, K.P.; Wu, R.; Harms, E.; Reiter, K.; et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J. Clin. Investig. 1998, 101, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Lübke, T.; Matthijs, G.; von Figura, K.; Körner, C. Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol. Cell. Biol. 2006, 26, 5615–5620. [Google Scholar] [CrossRef]
- DeRossi, C.; Bode, L.; Eklund, E.A.; Zhang, F.; Davis, J.A.; Westphal, V.; Wang, L.; Borowsky, A.D.; Freeze, H.H. Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J. Biol. Chem. 2006, 281, 5916–5927. [Google Scholar] [CrossRef]
- de Lonlay, P.; Seta, N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim. Biophys. Acta 2009, 1792, 841–843. [Google Scholar] [CrossRef]
- Pelletier, V.A.; Galéano, N.; Brochu, P.; Morin, C.L.; Weber, A.M.; Roy, C.C. Secretory diarrhea with protein-losing enteropathy, enterocolitis cystica superficialis, intestinal lymphangiectasia, and congenital hepatic fibrosis: A new syndrome. J. Pediatr. 1986, 108, 61–65. [Google Scholar] [CrossRef]
- Jaeken, J.; Matthijs, G.; Saudubray, J.M.; Dionisi-Vici, C.; Bertini, E.; de Lonlay, P.; Henri, H.; Carchon, H.; Schollen, E.; Van Schaftingen, E. Phosphomannose isomerase deficiency: A carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am. J. Hum. Genet. 1998, 62, 1535–1539. [Google Scholar] [CrossRef]
- Rymen, D.; Jaeken, J. Skin manifestations in CDG. J. Inherit. Metab. Dis. 2014, 37, 699–708. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Martín Hernández, E.; Vega Pajares, A.I.; Pérez González, B.; Ecay Crespo, M.J.; Leal Pérez, F.; Manzanares López-Manzanares, J.; Ugarte Pérez, M.; Pérez-Cerdá Silvestre, C. [Congenital disorder of glycosylation type 1b. Experience with mannose treatment]. An. Pediatr. 2008, 69, 358–365. [Google Scholar] [CrossRef]
- Mühlhausen, C.; Henneke, L.; Schlotawa, L.; Behme, D.; Grüneberg, M.; Gärtner, J.; Marquardt, T. Mannose phosphate isomerase deficiency-congenital disorder of glycosylation (MPI-CDG) with cerebral venous sinus thrombosis as first and only presenting symptom: A rare but treatable cause of thrombophilia. JIMD Rep. 2020, 55, 38–43. [Google Scholar] [CrossRef]
- Jaeken, J.; Lefeber, D.; Matthijs, G. Clinical utility gene card for: Phosphomannose isomerase deficiency. Eur. J. Hum. Genet. 2014, 22, 1153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Koning, T.J.; Dorland, L.; van Diggelen, O.P.; Boonman, A.M.; de Jong, G.J.; van Noort, W.L.; De Schryver, J.; Duran, M.; van den Berg, I.E.; Gerwig, G.J.; et al. A novel disorder of N-glycosylation due to phosphomannose isomerase deficiency. Biochem. Biophys. Res. Commun. 1998, 245, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Babovic-Vuksanovic, D.; Patterson, M.C.; Schwenk, W.F.; O’Brien, J.F.; Vockley, J.; Freeze, H.H.; Mehta, D.P.; Michels, V.V. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J. Pediatr. 1999, 135, 775–781. [Google Scholar] [CrossRef]
- Arndt, T.; Hackler, R.; Kleine, T.O.; Gressner, A.M. Validation by isoelectric focusing of the anion-exchange isotransferrin fractionation step involved in determination of carbohydrate-deficient transferrin by the CDTect assay. Clin. Chem. 1998, 44, 27–34. [Google Scholar] [CrossRef]
- Foo, Y.; Rosalki, S.B. Carbohydrate deficient transferrin measurement. Ann. Clin. Biochem. 1998, 35 Pt 3, 345–350. [Google Scholar] [CrossRef]
- Al Teneiji, A.; Bruun, T.U.; Sidky, S.; Cordeiro, D.; Cohn, R.D.; Mendoza-Londono, R.; Moharir, M.; Raiman, J.; Siriwardena, K.; Kyriakopoulou, L.; et al. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol. Genet. Metab. 2017, 120, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Quintana, E.; Montero, R.; Casado, M.; Navarro-Sastre, A.; Vilaseca, M.A.; Briones, P.; Artuch, R. Comparison between high performance liquid chromatography and capillary zone electrophoresis for the diagnosis of congenital disorders of glycosylation. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2009, 877, 2513–2518. [Google Scholar] [CrossRef]
- Crivellente, F.; Fracasso, G.; Valentini, R.; Manetto, G.; Riviera, A.P.; Tagliaro, F. Improved method for carbohydrate-deficient transferrin determination in human serum by capillary zone electrophoresis. J. Chromatogr. B. Biomed. Sci. Appl. 2000, 739, 81–93. [Google Scholar] [CrossRef]
- Helander, A.; Jaeken, J.; Matthijs, G.; Eggertsen, G. Asymptomatic phosphomannose isomerase deficiency (MPI-CDG) initially mistaken for excessive alcohol consumption. Clin. Chim. Acta 2014, 431, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; McClean, P.; Henderson, M.J.; Keir, D.G.; Worthington, V.C.; Imtiaz, F.; Schollen, E.; Matthijs, G.; Winchester, B.G. Successful treatment of carbohydrate deficient glycoprotein syndrome type 1b with oral mannose. Arch. Dis. Child 2001, 85, 339–340. [Google Scholar] [CrossRef]
- de Lonlay, P.; Cuer, M.; Vuillaumier-Barrot, S.; Beaune, G.; Castelnau, P.; Kretz, M.; Durand, G.; Saudubray, J.M.; Seta, N. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: A new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J. Pediatr. 1999, 135, 379–383. [Google Scholar] [CrossRef]
- Harms, H.K.; Zimmer, K.P.; Kurnik, K.; Bertele-Harms, R.M.; Weidinger, S.; Reiter, K. Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr. 2002, 91, 1065–1072. [Google Scholar] [CrossRef]
- Janssen, M.C.; de Kleine, R.H.; van den Berg, A.P.; Heijdra, Y.; van Scherpenzeel, M.; Lefeber, D.J.; Morava, E. Successful liver transplantation and long-term follow-up in a patient with MPI-CDG. Pediatrics 2014, 134, e279–e283. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H.; Eklund, E.A.; Ng, B.G.; Patterson, M.C. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012, 11, 453–466. [Google Scholar] [CrossRef]
- Li, L.B.; Chen, N.; Ramamoorthy, S.; Chi, L.; Cui, X.N.; Wang, L.C.; Reith, M.E. The role of N-glycosylation in function and surface trafficking of the human dopamine transporter. J. Biol. Chem. 2004, 279, 21012–21020. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Carchon, H. The carbohydrate-deficient glycoprotein syndromes: An overview. J. Inherit. Metab. Dis. 1993, 16, 813–820. [Google Scholar] [CrossRef]
- Barone, R.; Carrozzi, M.; Parini, R.; Battini, R.; Martinelli, D.; Elia, M.; Spada, M.; Lilliu, F.; Ciana, G.; Burlina, A.; et al. A nationwide survey of PMM2-CDG in Italy: High frequency of a mild neurological variant associated with the L32R mutation. J. Neurol. 2015, 262, 154–164. [Google Scholar] [CrossRef]
- Pérez, B.; Briones, P.; Quelhas, D.; Artuch, R.; Vega, A.I.; Quintana, E.; Gort, L.; Ecay, M.J.; Matthijs, G.; Ugarte, M.; et al. The molecular landscape of phosphomannose mutase deficiency in iberian peninsula: Identification of 15 population-specific mutations. JIMD Rep. 2011, 1, 117–123. [Google Scholar] [PubMed]
- Coman, D.; McGill, J.; MacDonald, R.; Morris, D.; Klingberg, S.; Jaeken, J.; Appleton, D. Congenital disorder of glycosylation type 1a: Three siblings with a mild neurological phenotype. J. Clin. Neurosci. 2007, 14, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Tajima, G.; Ono, H.; Kobayashi, M. Different neuroradiological findings during two stroke-like episodes in a patient with a congenital disorder of glycosylation type Ia. Brain Dev. 2009, 31, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Schollen, E.; Pardon, E.; Veiga-Da-Cunha, M.; Jaeken, J.; Cassiman, J.J.; Van Schaftingen, E. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type, I. syndrome (Jaeken syndrome). Nat. Genet. 1997, 16, 88–92. [Google Scholar] [CrossRef]
- Funke, S.; Gardeitchik, T.; Kouwenberg, D.; Mohamed, M.; Wortmann, S.B.; Korsch, E.; Adamowicz, M.; Al-Gazali, L.; Wevers, R.A.; Horvath, A.; et al. Perinatal and early infantile symptoms in congenital disorders of glycosylation. Am. J. Med. Genet. A. 2013, 161a, 578–584. [Google Scholar] [CrossRef]
- Resende, C.; Carvalho, C.; Alegria, A.; Oliveira, D.; Quelhas, D.; Bandeira, A.; Proença, E. Congenital disorders of glycosylation with neonatal presentation. BMJ Case Rep. 2014, 2014, bcr2013010037. [Google Scholar] [CrossRef]
- Serrano, M.; de Diego, V.; Muchart, J.; Cuadras, D.; Felipe, A.; Macaya, A.; Velázquez, R.; Poo, M.P.; Fons, C.; O’Callaghan, M.M.; et al. Phosphomannomutase deficiency (PMM2-CDG): Ataxia and cerebellar assessment. Orphanet J. Rare Dis. 2015, 10, 138. [Google Scholar] [CrossRef]
- Stibler, H.; Blennow, G.; Kristiansson, B.; Lindehammer, H.; Hagberg, B. Carbohydrate-deficient glycoprotein syndrome: Clinical expression in adults with a new metabolic disease. J. Neurol. Neurosurg. Psychiatry 1994, 57, 552–556. [Google Scholar] [CrossRef]
- Monin, M.L.; Mignot, C.; De Lonlay, P.; Héron, B.; Masurel, A.; Mathieu-Dramard, M.; Lenaerts, C.; Thauvin, C.; Gérard, M.; Roze, E.; et al. 29 French adult patients with PMM2-congenital disorder of glycosylation: Outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J. Rare Dis. 2014, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Shanti, B.; Silink, M.; Bhattacharya, K.; Howard, N.J.; Carpenter, K.; Fietz, M.; Clayton, P.; Christodoulou, J. Congenital disorder of glycosylation type Ia: Heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S241–S251. [Google Scholar] [CrossRef]
- Krasnewich, D.; O’Brien, K.; Sparks, S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG-Ia). Am. J. Med. Genet. C Semin. Med. Genet. 2007, 145c, 302–306. [Google Scholar] [CrossRef]
- Martinez-Monseny, A.; Cuadras, D.; Bolasell, M.; Muchart, J.; Arjona, C.; Borregan, M.; Algrabli, A.; Montero, R.; Artuch, R.; Velázquez-Fragua, R.; et al. From gestalt to gene: Early predictive dysmorphic features of PMM2-CDG. J. Med. Genet. 2019, 56, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.C.; Marquardt, T.; Lausch, E.; Fuchs, H.; Thiel, C.; Sutter, M.; Schumann, A.; Hannibal, L.; Spiekerkoetter, U. Unsuccessful intravenous D-mannose treatment in PMM2-CDG. Orphanet. J. Rare Dis. 2019, 14, 231. [Google Scholar] [CrossRef]
- Rush, J.S.; Panneerselvam, K.; Waechter, C.J.; Freeze, H.H. Mannose supplementation corrects GDP-mannose deficiency in cultured fibroblasts from some patients with Congenital Disorders of Glycosylation (CDG). Glycobiology 2000, 10, 829–835. [Google Scholar] [CrossRef]
- Schneider, A.; Thiel, C.; Rindermann, J.; De Rossi, C.; Popovici, D.; Hoffmann, G.F.; Gröne, H.J.; Körner, C. Successful prenatal mannose treatment for congenital disorder of glycosylation-Ia in mice. Nat. Med. 2011, 18, 71–73. [Google Scholar] [CrossRef]
- Kjaergaard, S.; Kristiansson, B.; Stibler, H.; Freeze, H.H.; Schwartz, M.; Martinsson, T.; Skovby, F. Failure of short-term mannose therapy of patients with carbohydrate-deficient glycoprotein syndrome type 1A. Acta Paediatr. 1998, 87, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Liguori, L.; Allocca, M.; Andreotti, G.; Cubellis, M.V. β-Glucose-1,6-Bisphosphate Stabilizes Pathological Phophomannomutase2 Mutants In Vitro and Represents a Lead Compound to Develop Pharmacological Chaperones for the Most Common Disorder of Glycosylation, PMM2-CDG. Int. J. Mol. Sci. 2019, 20, 4164. [Google Scholar] [CrossRef]
- Regal, L.; van Hasselt, P.M.; Foulquier, F.; Cuppen, I.; Prinsen, H.; Jansen, K.; Keldermans, L.; De Meirleir, L.; Matthijs, G.; Jaeken, J. ALG11-CDG: Three novel mutations and further characterization of the phenotype. Mol. Genet. Metab. Rep. 2015, 2, 16–19. [Google Scholar] [CrossRef]
- Rind, N.; Schmeiser, V.; Thiel, C.; Absmanner, B.; Lübbehusen, J.; Hocks, J.; Apeshiotis, N.; Wilichowski, E.; Lehle, L.; Körner, C. A severe human metabolic disease caused by deficiency of the endoplasmatic mannosyltransferase hALG11 leads to congenital disorder of glycosylation-Ip. Hum. Mol. Genet. 2010, 19, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Rind, N.; Popovici, D.; Hoffmann, G.F.; Hanson, K.; Conway, R.L.; Adamski, C.R.; Butler, E.; Scanlon, R.; Lambert, M.; et al. Improved diagnostics lead to identification of three new patients with congenital disorder of glycosylation-Ip. Hum. Mutat. 2012, 33, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Gawor, M.; Prószyński, T.J. The molecular cross talk of the dystrophin-glycoprotein complex. Ann. N. Y. Acad. Sci. 2018, 1412, 62–72. [Google Scholar] [CrossRef]
- Roomi, M.W.; Ishaque, A.; Khan, N.R.; Eylar, E.H. The P.O. protein. The major glycoprotein of peripheral nerve myelin. Biochim. Biophys. Acta 1978, 536, 112–121. [Google Scholar] [CrossRef]
- Haanpää, M.K.; Ng, B.G.; Gallant, N.M.; Singh, K.E.; Brown, C.; Kimonis, V.; Freeze, H.H.; Muller, E.A. 2nd: ALG11-CDG syndrome: Expanding the phenotype. Am. J. Med. Genet. A 2019, 179, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Sparks, S.E.; Krasnewich, D.M. Congenital disorders of N-linked glycosylation and multiple pathway overview. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Sone, H.; Shimano, H.; Ebinuma, H.; Takahashi, A.; Yano, Y.; Iida, K.T.; Suzuki, H.; Toyoshima, H.; Kawakami, Y.; Okuda, Y.; et al. Physiological changes in circulating mannose levels in normal, glucose-intolerant, and diabetic subjects. Metabolism 2003, 52, 1019–1027. [Google Scholar] [CrossRef]
- Gonzalez, P.S.; O’Prey, J.; Cardaci, S.; Barthet, V.J.A.; Sakamaki, J.I.; Beaumatin, F.; Roseweir, A.; Gay, D.M.; Mackay, G.; Malviya, G.; et al. Mannose impairs tumour growth and enhances chemotherapy. Nature 2018, 563, 719–723. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, S.; He, B. Mannose shows antitumour properties against lung cancer via inhibiting proliferation, promoting cisplatin-mediated apoptosis and reducing metastasis. Mol. Med. Rep. 2020, 22, 2957–2965. [Google Scholar] [CrossRef]
- Kranjčec, B.; Papeš, D.; Altarac, S. D-mannose powder for prophylaxis of recurrent urinary tract infections in women: A randomized clinical trial. World. J. Urol. 2014, 32, 79–84. [Google Scholar] [CrossRef]
- Domenici, L.; Monti, M.; Bracchi, C.; Giorgini, M.; Colagiovanni, V.; Muzii, L.; Benedetti Panici, P. D-mannose: A promising support for acute urinary tract infections in women. A pilot study. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2920–2925. [Google Scholar]
- Sharma, V.; Smolin, J.; Nayak, J.; Ayala, J.E.; Scott, D.A.; Peterson, S.N.; Freeze, H.H. Mannose Alters Gut Microbiome, Prevents Diet-Induced Obesity, and Improves Host Metabolism. Cell Rep. 2018, 24, 3087–3098. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Metabolite | Disorder | Enzyme Deficiency | Affected Organs/Organ System | Clinical Manifestations | Treatment |
|---|---|---|---|---|---|
| Fructose | Essential Fructosuria | KHK | Liver, circulatory system | Fructosuria | No treatment necessary |
| Hereditary Fructose Intolerance | ALDOB | Liver, kidney | Hypoglycemia, nausea, vomiting, abdominal pain upon fructose consumption, aminoaciduria, fructosuria | Dietary restriction of fructose, sorbitol, and sucrose | |
| FBPase Deficiency | FBP1 | Liver, brain, heart | Severe hypoglycemia, hepatomegaly, acidoketosis, episodic acute crises of hyperventilation, coma, seizures, irritability, tachycardia | Dietary restriction of fructose, oral/IV glucose administration, avoidance of fasting, supplementing water with cornstarch to prevent nocturnal hypoglycemia | |
| Mannose | MPI-CDG | MPI | Digestive tract | Liver fibrosis, hyperglycemia, protein-loss enteropathy, failure to thrive | Oral mannose |
| PMM2-CDG | PMM2 | Central nervous system, circulatory system | Infantile multisystem: developmental delays, failure to thrive, and neuropathy Late-infantile and childhood ataxia-intellectual disability: language and motor delays, stroke-like episodes Adult stable disability: premature aging, abnormal sexual development, and increased risk of deep-vein thrombosis | Occupational therapy, maintenance of healthy blood glucose | |
| ALG11-CDG | ALG11 | Central nervous system, circulatory system, endocrine system | Developmental and language delay, axial hypotonia, seizures, dysmorphic facial features, inverted nipples | Plasma infusions, hormonal regimens |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lieu, E.L.; Kelekar, N.; Bhalla, P.; Kim, J. Fructose and Mannose in Inborn Errors of Metabolism and Cancer. Metabolites 2021, 11, 479. https://doi.org/10.3390/metabo11080479
Lieu EL, Kelekar N, Bhalla P, Kim J. Fructose and Mannose in Inborn Errors of Metabolism and Cancer. Metabolites. 2021; 11(8):479. https://doi.org/10.3390/metabo11080479
Chicago/Turabian StyleLieu, Elizabeth L., Neil Kelekar, Pratibha Bhalla, and Jiyeon Kim. 2021. "Fructose and Mannose in Inborn Errors of Metabolism and Cancer" Metabolites 11, no. 8: 479. https://doi.org/10.3390/metabo11080479
APA StyleLieu, E. L., Kelekar, N., Bhalla, P., & Kim, J. (2021). Fructose and Mannose in Inborn Errors of Metabolism and Cancer. Metabolites, 11(8), 479. https://doi.org/10.3390/metabo11080479