
Metformin Decreases 2-HG Production through the MYC-PHGDH Pathway in Suppressing Breast Cancer Cell Proliferation


Abstract
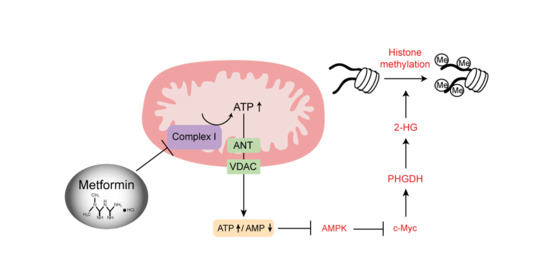
:
1. Introduction
2. Results
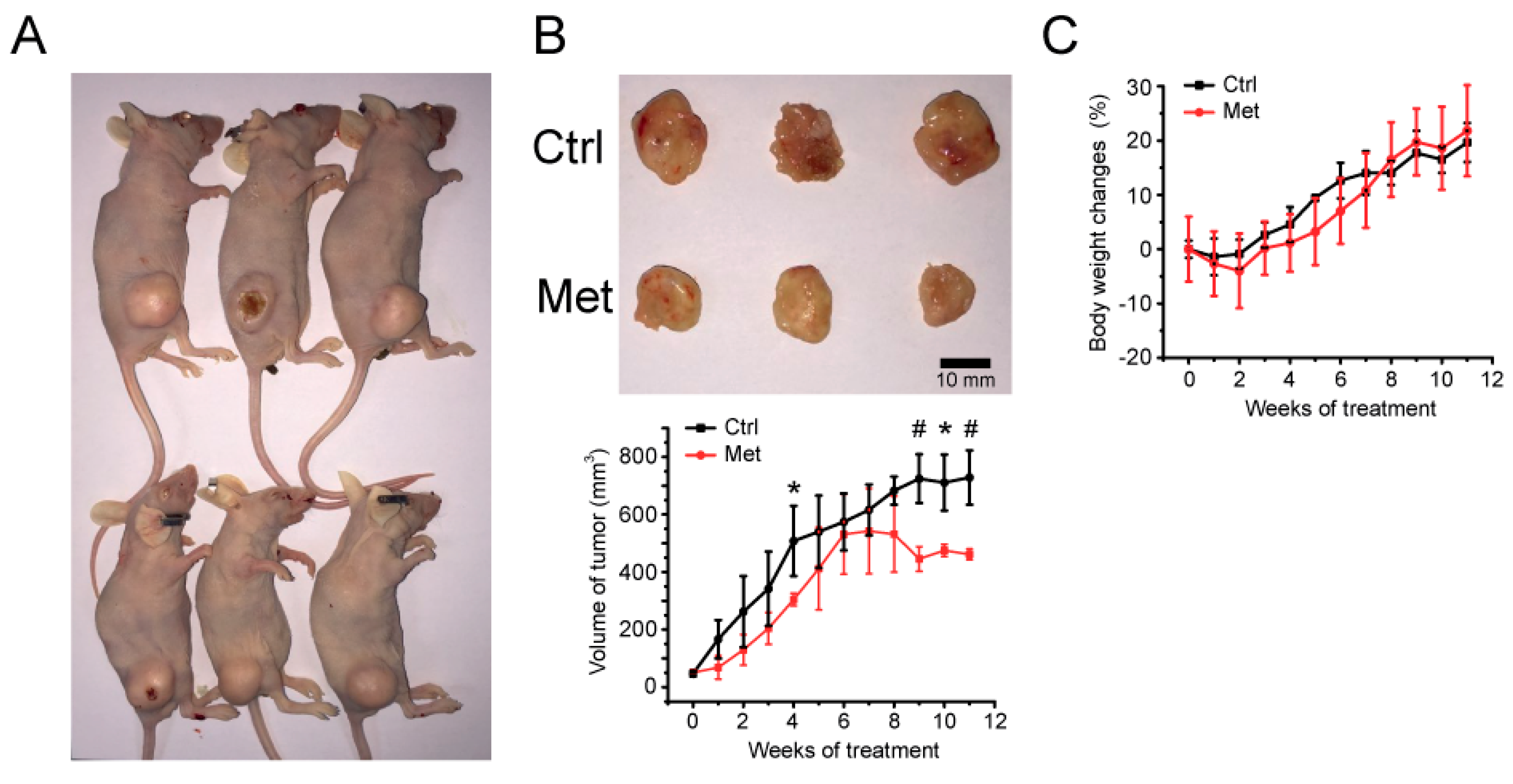
2.1. Xenograft Study on Metformin’s Cancer Prevention Effect
2.2. Metformin’s Inhibitory Activity against Breast Cancer Cells
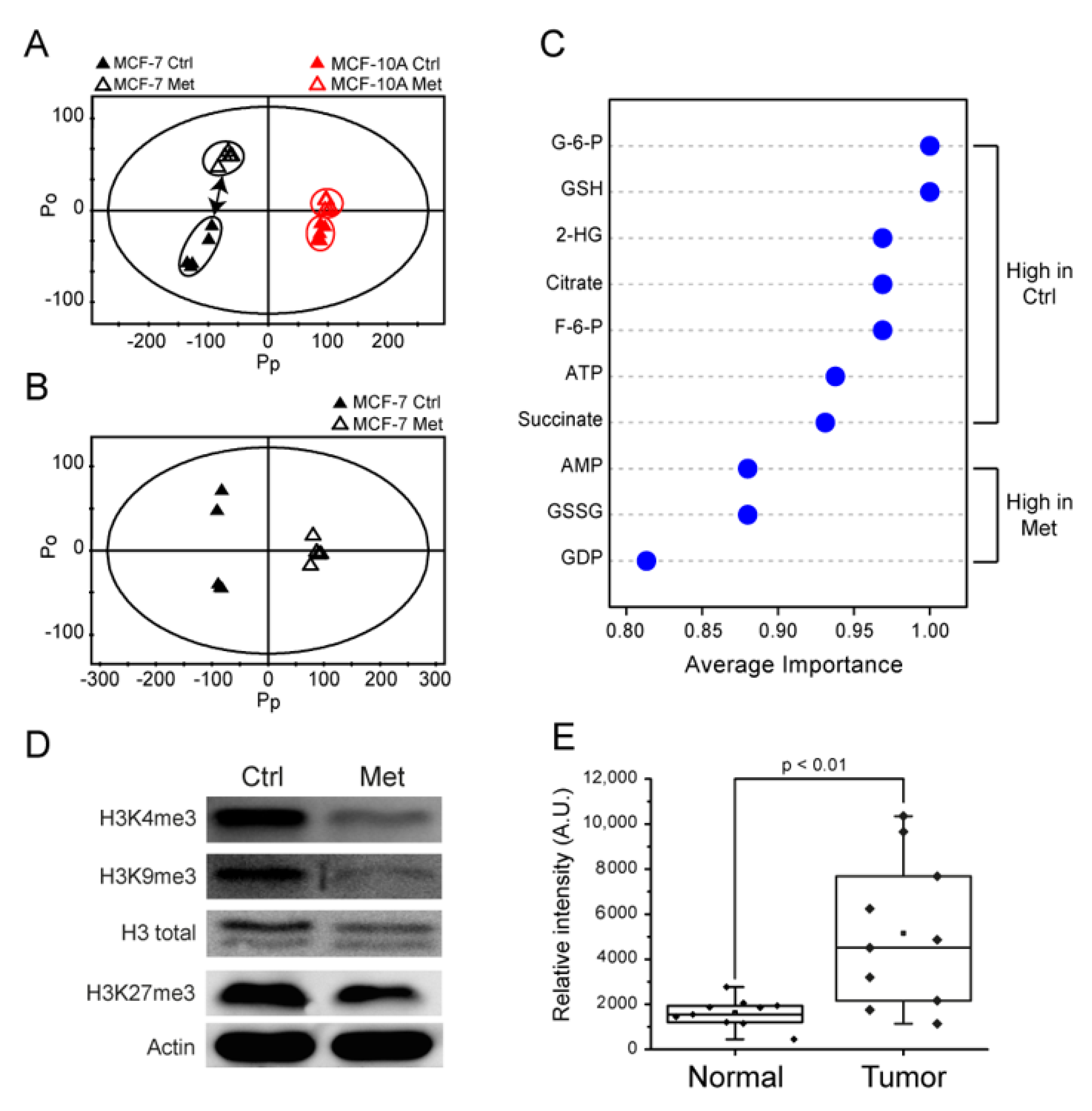
2.3. Metformin Induces Metabolomic Changes and Decreases 2-HG Level in MCF-7 Cells
2.4. Metformin Changes Histone Methylation Status of MCF-7 Cells
2.5. The 2-HG Level Is Higher in Tumor Tissue Samples
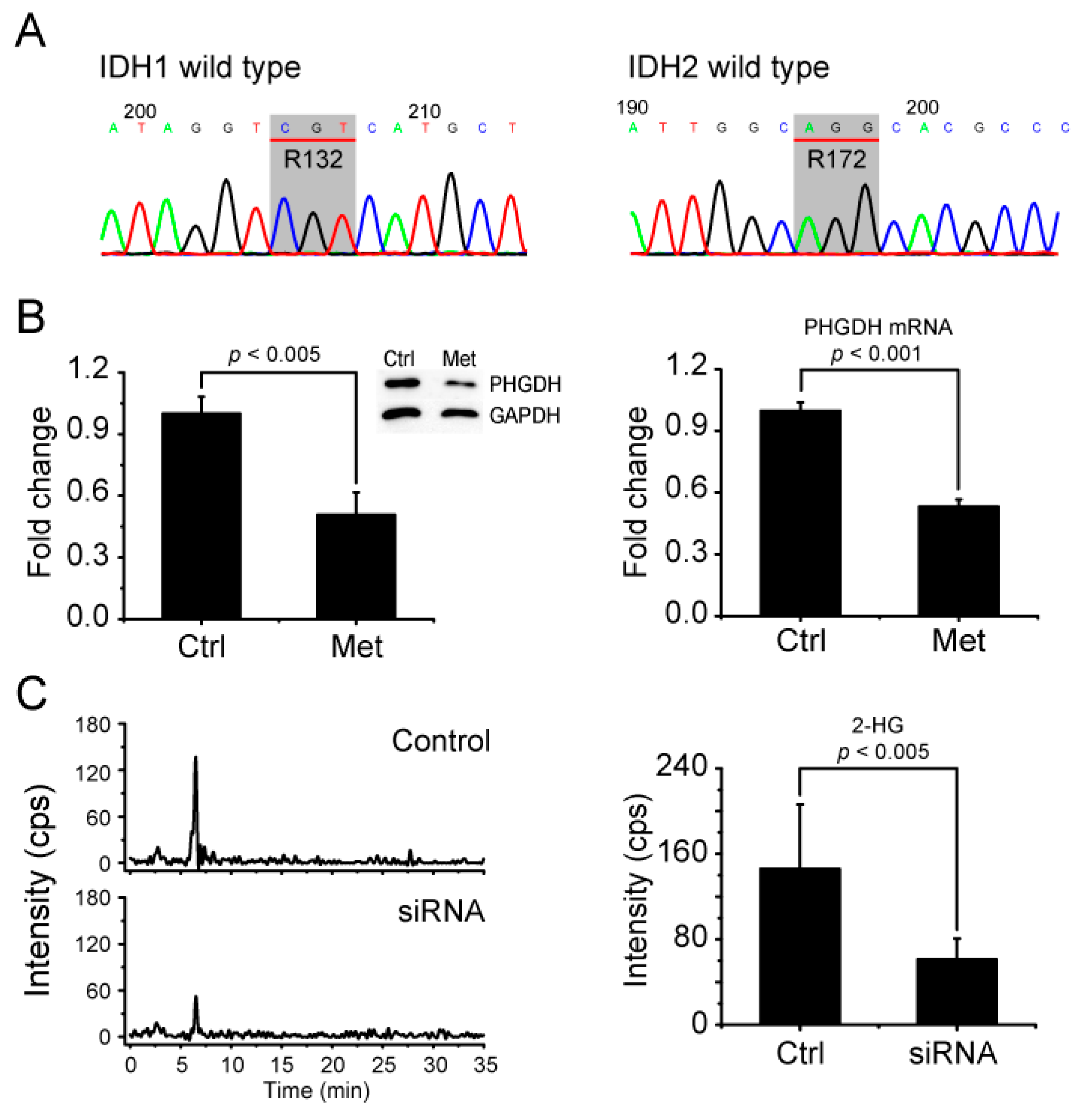
2.6. Metformin Reduces 2-HG Level through Downregulation of PHGDH
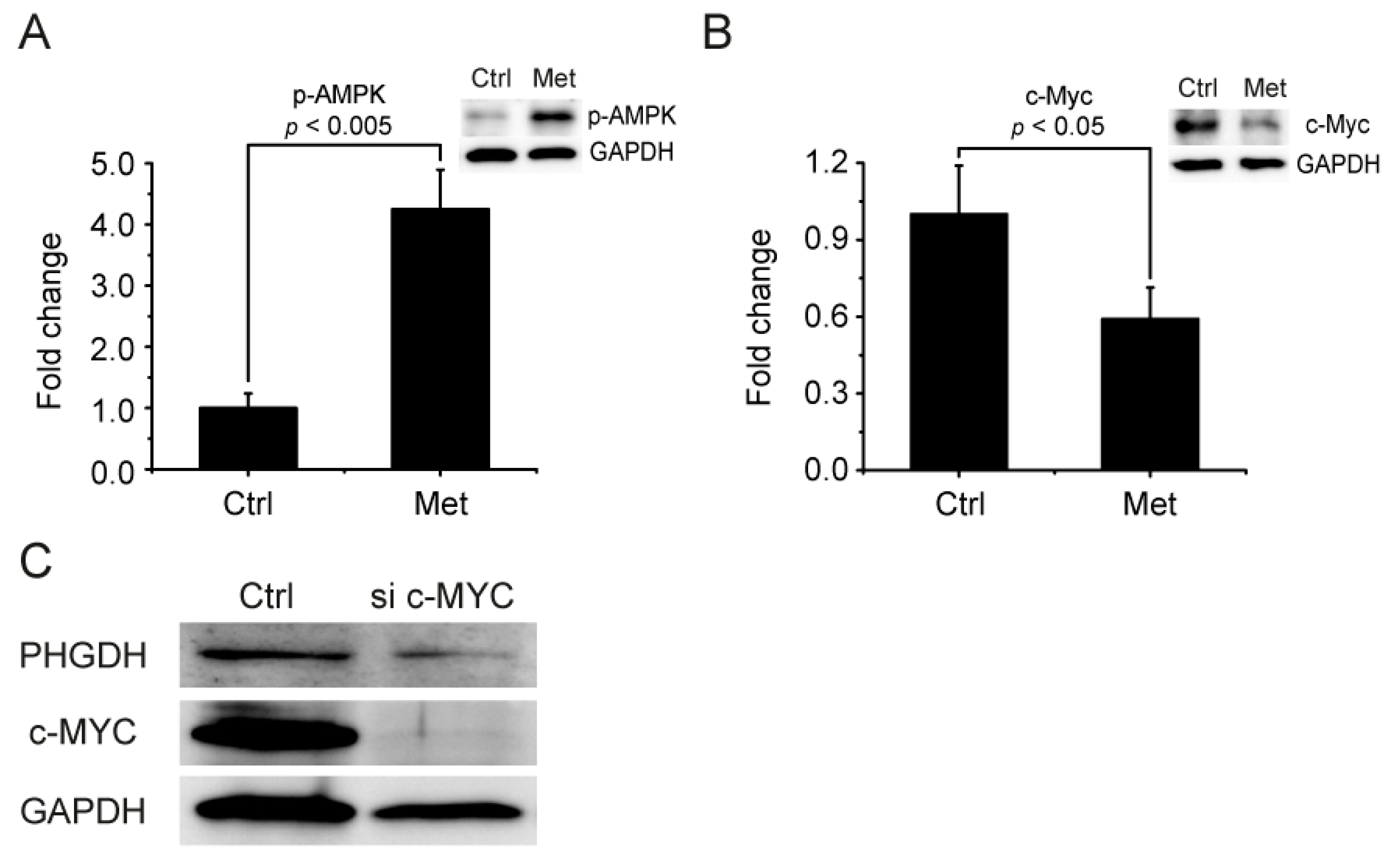
2.7. AMPK-MYC Pathway Is Implicated in the Regulation of PHGDH-2HG by Metformin
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell lines and Metformin Treatment
4.3. Xenograft Mouse Model with MCF-7 Cells
4.4. Metabolomic Analysis and Metabolite Measurements
4.5. ROS Measurement
4.6. Quantitative Real-Time PCR and Western Blot Analysis
4.7. 2-HG Quantification from Human Breast Tissue Samples
4.8. DNA and Histone Methylation Analysis
4.9. PHGDH Knockdown with siRNA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2012, 35, 731–737. [Google Scholar]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.-Y.; Cheng, J.-T.; Yu, E.; Wu, T.-J. Metformin increases insulin sensitivity and plasma β-endorphin in human subjects. Horm. Metab. Res. 2006, 38, 106–111. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143. [Google Scholar] [CrossRef]
- Spiering, M.J. The mystery of metformin. J. Biol. Chem. 2019, 294, 6689–6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240. [Google Scholar] [CrossRef]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, T.I.; Jeon, S.M.; Hong, S.P.; Cheon, J.H.; Kim, W.H. The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. Int. J. Cancer 2012, 131, 752–759. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef] [Green Version]
- Suissa, S.; Azoulay, L. Metformin and cancer: Mounting evidence against an association. Diabetes Care 2014, 37, 1786–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Frasca, F. IGF and insulin receptor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathot, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K. Phase II clinical trial of metformin as a cancer stem cell–targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Mullarky, E.; Mattaini, K.R.; Vander Heiden, M.G.; Cantley, L.C.; Locasale, J.W. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 2011, 24, 1112–1115. [Google Scholar] [CrossRef]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Teng, X.; Liu, L.; Mattaini, K.R.; Looper, R.E.; Vander Heiden, M.G.; Rabinowitz, J.D. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem. Biol. 2015, 10, 510–516. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Chen, W.L.; Li, J.M.; Wu, S.F.; Chen, T.L.; Zhu, Y.M.; Zhang, W.N.; Li, Y.; Qiu, Y.P.; Zhao, A.H.; et al. Prognostic significance of 2-hydroxyglutarate levels in acute myeloid leukemia in China. Proc. Natl. Acad. Sci. USA 2013, 110, 17017–17022. [Google Scholar] [CrossRef] [Green Version]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathe, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef] [Green Version]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Deng, X.S.; Alimova, I.N.; Lind, S.E.; Thor, A.D. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 2009, 8, 2031–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, A.; Markert, E.K.; Oltvai, Z.N. Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. PLoS ONE 2011, 6, e25881. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.C.; Song, L.B.; Wan, Q.F.; Wu, G.W.; Li, X.H.; Wang, Y.H.; Wang, J.; Liu, Z.J.; Zhong, X.Y.; He, X.P.; et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat. Commun. 2012, 3, 865. [Google Scholar] [CrossRef]
- Akinyeke, T.; Matsumura, S.; Wang, X.; Wu, Y.; Schalfer, E.D.; Saxena, A.; Yan, W.; Logan, S.K.; Li, X. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis 2013, 34, 2823–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Barco, S.; Vazquez-Martin, A.; Cufi, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef] [Green Version]
- Ben Sahra, I.; Le Marchand-Brustel, Y.; Tanti, J.F.; Bost, F. Metformin in cancer therapy: A new perspective for an old antidiabetic drug? Mol. Cancer Ther. 2010, 9, 1092–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, A.; Di Gennaro, E.; Bruzzese, F.; Avallone, A.; Budillon, A. New perspective for an old antidiabetic drug: Metformin as anticancer agent. In Advances in Nutrition and Cancer; Springer: Berlin/Heidelberg, Germany, 2014; pp. 355–376. [Google Scholar]
- Cuyas, E.; Fernandez-Arroyo, S.; Corominas-Faja, B.; Rodriguez-Gallego, E.; Bosch-Barrera, J.; Martin-Castillo, B.; De Llorens, R.; Joven, J.; Menendez, J.A. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget 2015, 6, 12279–12296. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, P.J.; Ligibel, J.A.; Stambolic, V. Metformin in breast cancer: Time for action. J. Clin. Oncol. 2009, 27, 3271–3273. [Google Scholar] [CrossRef]
- Smolková, K.; Dvořák, A.; Zelenka, J.; Vítek, L.; Ježek, P. Reductive carboxylation and 2-hydroxyglutarate formation by wild–type IDH2 in breast carcinoma cells. Int. J. Biochem. Cell Biol. 2015. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016, 76, 4430–4442. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Xu, W.J.; Jin, X.; Oh, S.; Phan, C.H.D.; Song, J.; Lee, S.K.; Park, S. The roles of IP 3 receptor in energy metabolic pathways and reactive oxygen species homeostasis revealed by metabolomic and biochemical studies. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 2937–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Yang, H.J.; An, Y.J.; Kim, J.M.; Lee, D.H.; Jin, X.; Park, S.W.; Min, K.J.; Park, S. Enhanced phase II detoxification contributes to beneficial effects of dietary restriction as revealed by multi-platform metabolomics studies. Mol. Cell. Proteom. 2013, 12, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Shen, L.; Cheng, A.S.; Ahmed, S.; Boumber, Y.; Charo, C.; Yamochi, T.; Urano, T.; Furukawa, K.; Kwabi-Addo, B.; et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 2008, 40, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [Green Version]
- Han, T.K.; Proctor, W.R.; Costales, C.L.; Cai, H.; Everett, R.S.; Thakker, D.R. Four cation-selective transporters contribute to apical uptake and accumulation of metformin in Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 2015, 352, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Pawlikowski, B.; Schlessinger, A.; More, S.S.; Stryke, D.; Johns, S.J.; Portman, M.A.; Chen, E.; Ferrin, T.E.; Sali, A.; et al. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenet. Genom. 2010, 20, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algire, C.; Zakikhani, M.; Blouin, M.J.; Shuai, J.H.; Pollak, M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr. Relat. Cancer 2008, 15, 833–839. [Google Scholar] [CrossRef]
- Dowling, R.J.; Goodwin, P.J.; Stambolic, V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, P.J.; Pritchard, K.I.; Ennis, M.; Clemons, M.; Graham, M.; Fantus, I.G. Insulin-lowering effects of metformin in women with early breast cancer. Clin. Breast Cancer 2008, 8, 501–505. [Google Scholar] [CrossRef]
- Lega, I.C.; Austin, P.C.; Gruneir, A.; Goodwin, P.J.; Rochon, P.A.; Lipscombe, L.L. Association Between Metformin Therapy and Mortality After Breast Cancer A population-based study. Diabetes Care 2013, 36, 3018–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kwon, H.; Lee, J.W.; Kim, H.J.; Lee, S.B.; Park, H.S.; Sohn, G.; Lee, Y.; Koh, B.S.; Yu, J.H.; et al. Metformin increases survival in hormone receptor-positive, HER2-positive breast cancer patients with diabetes. Breast Cancer Res. 2015, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Dubois, V.; Dasnois, L.; Lebtahi, K.; Collot, F.; Heylen, N.; Havaux, N.; Fernandez, A.M.; Lobl, T.J.; Oliyai, C.; Nieder, M.; et al. CPI-0004Na, a new extracellularly tumor-activated prodrug of doxorubicin: In vivo toxicity, activity, and tissue distribution confirm tumor cell selectivity. Cancer Res. 2002, 62, 2327–2331. [Google Scholar] [PubMed]
- Wen, H.; Cho, H.R.; Yun, T.; Kim, H.; Park, C.K.; Lee, S.H.; Choi, S.H.; Park, S. Metabolomic comparison between cells over-expressing isocitrate dehydrogenase 1 and 2 mutants and the effects of an inhibitor on the metabolism. J. Neurochem. 2015, 132, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.; Fernandes, E.; Lima, J.L.F.C. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Fold Change (%) | p Value | |
|---|---|---|---|
| Increased | Acetate | 43.54 | <0.001 |
| Alanine | 18.65 | <0.001 | |
| AMP | 46.17 | <0.0053 | |
| Creatine | 196.27 | <0.001 | |
| GDP | 38.50 | <0.01 | |
| Glutamine | 87.94 | <0.001 | |
| GSSG | 153.39 | <0.001 | |
| Isoleucine | 78.43 | <0.001 | |
| Leucine | 67.51 | <0.001 | |
| Decreased | ATP | 47.72 | <0.05 |
| Citrate | 55.23 | <0.001 | |
| F-6-P | 66.49 | <0.001 | |
| GSH | 17.52 | <0.005 | |
| G-6-P | 69.91 | <0.001 | |
| Lactate | 19.37 | <0.05 | |
| O-Phosphocholine | 33.08 | <0.001 | |
| Succinate | 84.07 | <0.05 | |
| 2-HG | 68.85 | <0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.; Cho, Y.; Chang, M.; Park, S.; Kwon, H. Metformin Decreases 2-HG Production through the MYC-PHGDH Pathway in Suppressing Breast Cancer Cell Proliferation. Metabolites 2021, 11, 480. https://doi.org/10.3390/metabo11080480
Oh S, Cho Y, Chang M, Park S, Kwon H. Metformin Decreases 2-HG Production through the MYC-PHGDH Pathway in Suppressing Breast Cancer Cell Proliferation. Metabolites. 2021; 11(8):480. https://doi.org/10.3390/metabo11080480
Chicago/Turabian StyleOh, Sehyun, Youngup Cho, Minsun Chang, Sunghyouk Park, and Hyuknam Kwon. 2021. "Metformin Decreases 2-HG Production through the MYC-PHGDH Pathway in Suppressing Breast Cancer Cell Proliferation" Metabolites 11, no. 8: 480. https://doi.org/10.3390/metabo11080480
APA StyleOh, S., Cho, Y., Chang, M., Park, S., & Kwon, H. (2021). Metformin Decreases 2-HG Production through the MYC-PHGDH Pathway in Suppressing Breast Cancer Cell Proliferation. Metabolites, 11(8), 480. https://doi.org/10.3390/metabo11080480