
Cryptosporidiosis Modulates the Gut Microbiome and Metabolism in a Murine Infection Model

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Mouse Strain Selection for Multi-Omics Studies
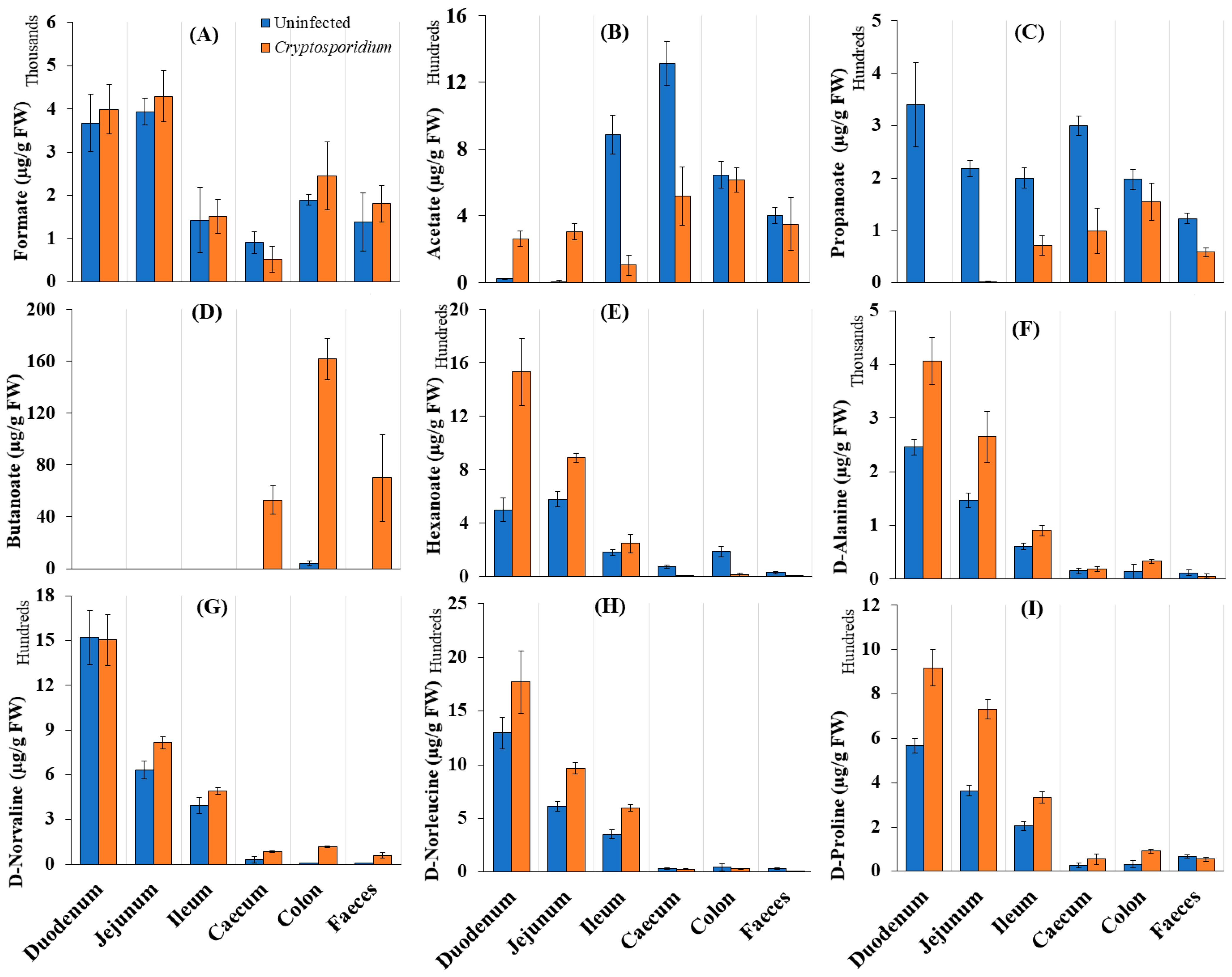
2.2. Gut Metabolism and Major Metabolic Pathways
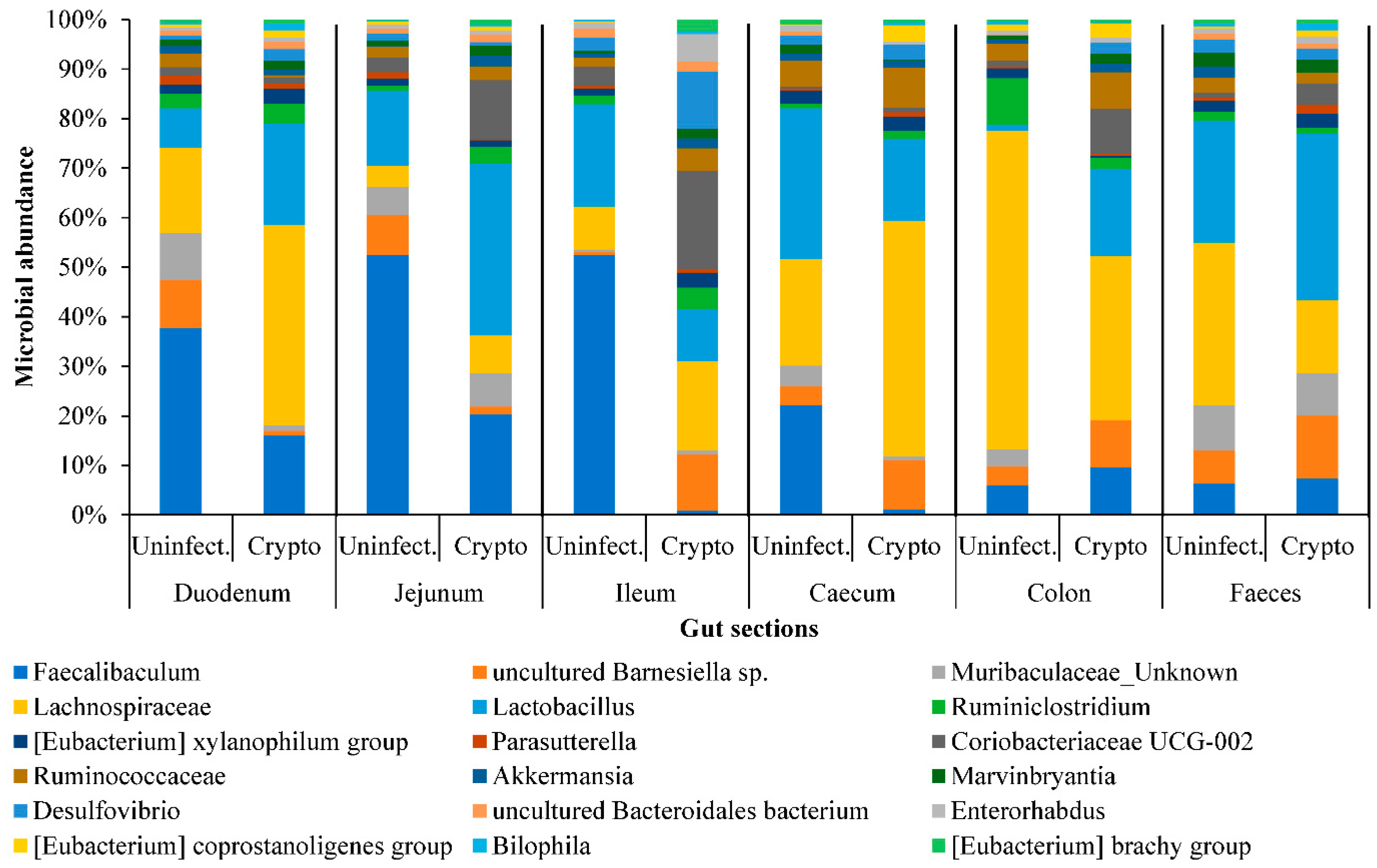
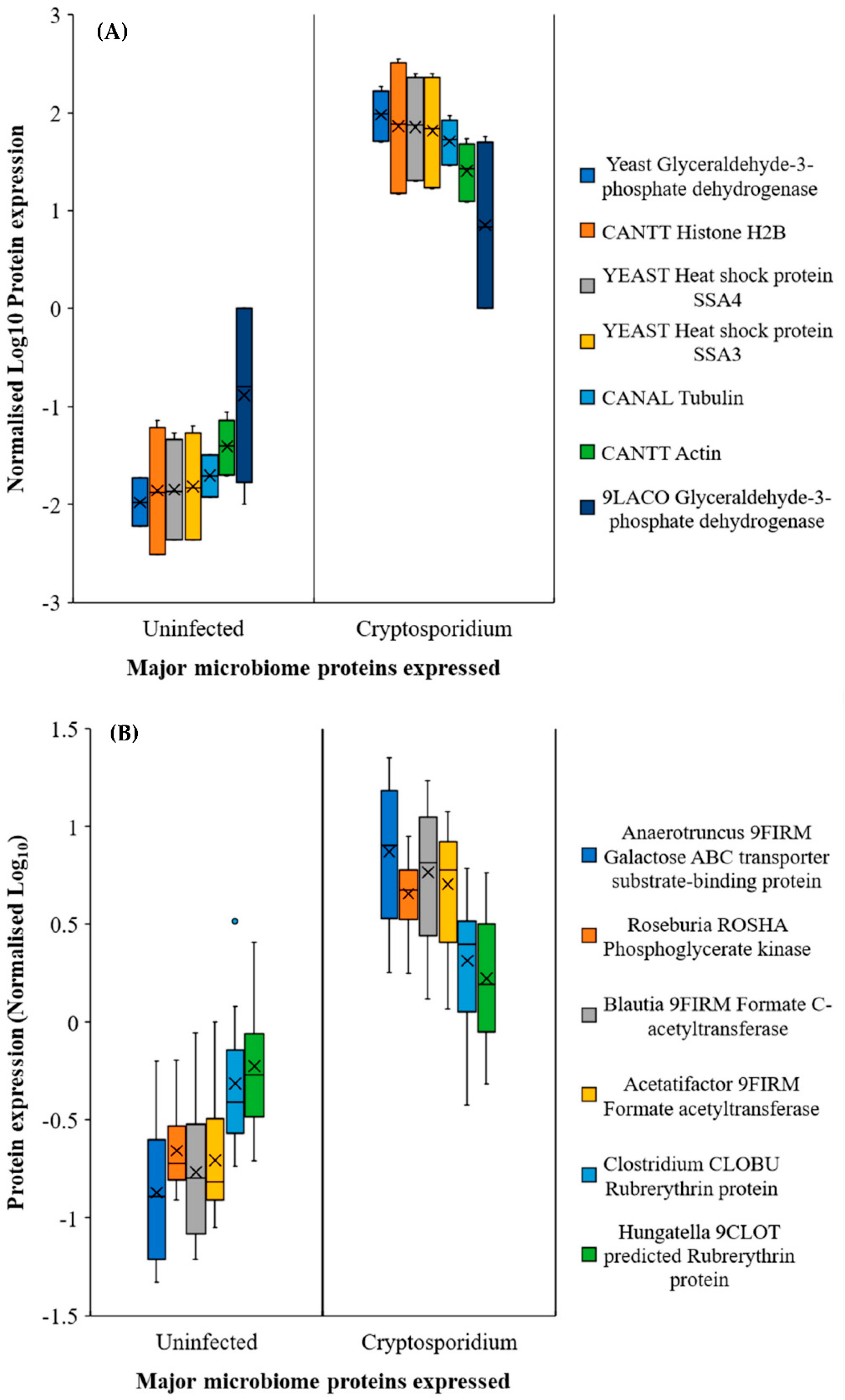
2.3. Gut Microbiome Response and Altered Energy Metabolic Pathways during Cryptosporidiosis
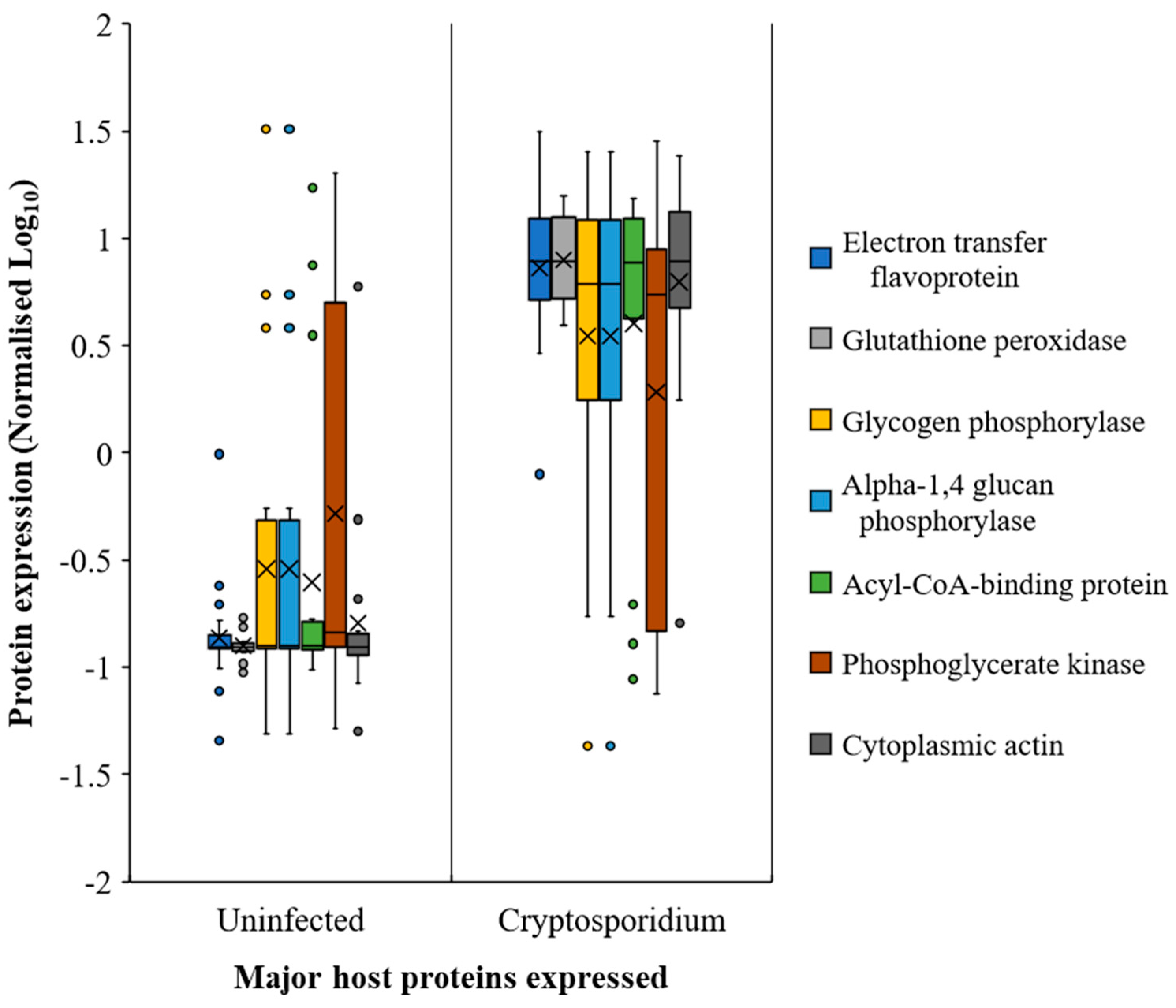
2.4. Host Response in the Gut during Cryptosporidiosis
2.5. Extra-Intestinal Effects of Cryptosporidiosis
3. Discussion
3.1. Cryptosporidiosis Dynamics in the Gut
3.2. Extra-Intestinal Effects of Cryptosporidiosis
4. Materials and Methods
4.1. Animal Ethics and Husbandry
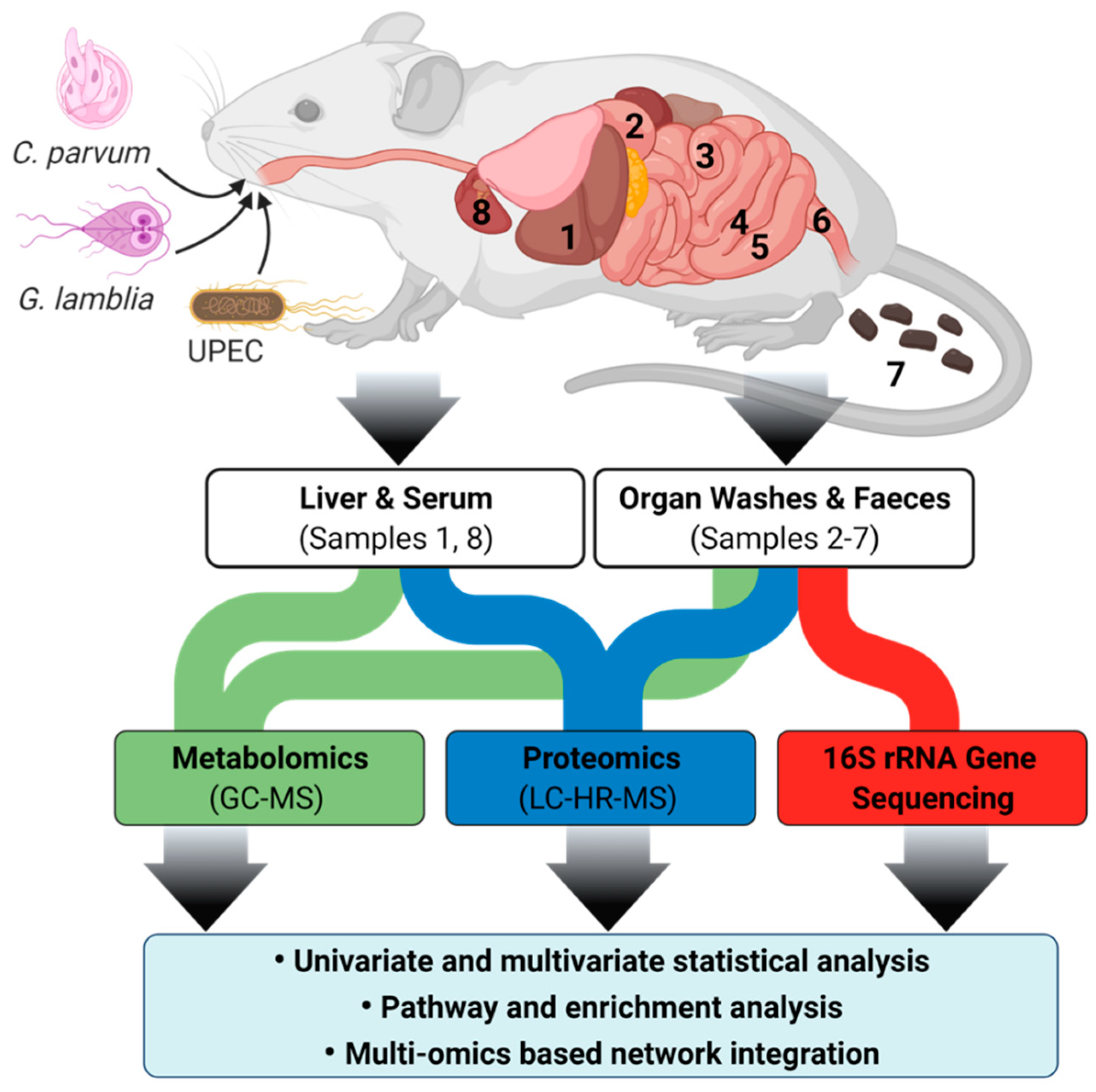
4.2. Mouse Infection Model
4.3. Untargeted Metabolomics by Gas Chromatography-Mass Spectrometry (GC-MS)
4.4. Metaproteome Extraction and Proteome Analysis
4.5. Genomic Extraction, Analysis and Processing
4.6. Multi-Omics Integration and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kotloff, K.L.; Nasrin, D.; Blackwelder, W.C.; Wu, Y.; Farag, T.; Panchalingham, S.; Sow, S.O.; Sur, D.; Zaidi, A.K.M.; Faruque, A.S.G.; et al. The incidence, aetiology, and adverse clinical consequences of less severe diarrhoeal episodes among infants and children residing in low-income and middle-income countries: A 12-month case-control study as a follow-on to the Global Enteric Multicenter Study (GEMS). Lancet Glob. Health 2019, 7, e568–e584. [Google Scholar] [CrossRef]
- Khalil, I.A.; Troeger, C.; Rao, P.C.; Blacker, B.F.; Brown, A.; Brewer, T.G.; Colombara, D.V.; De Hostos, E.L.; Engmann, C.; Guerrant, R.L.; et al. Morbidity, mortality, and long-term consequences associated with diarrhoea from Cryptosporidium infection in children younger than 5 years: A meta-analyses study. Lancet Glob. Health 2018, 6, e758–e768. [Google Scholar] [CrossRef]
- Certad, G.; Viscogliosi, E.; Chabé, M.; Cacciò, S.M. Pathogenic Mechanisms of Cryptosporidium and Giardia. Trends Parasitol. 2017, 33, 561–576. [Google Scholar] [CrossRef]
- Priyamvada, S.; Kumar, A.; Anbazhagan, A.N.; Jayawardena, D.; Alrefai, W.A.; Dudeja, P.K.; Bortahkur, A. Cryptosporidium parvum infection induces autophagy in intestinal epithelial cells. Gastroenterology 2019, 156, e13298. [Google Scholar] [CrossRef]
- Alcock, F.; Webb, C.T.; Dolezal, P.; Hewitt, V.; Shingu-Vasquez, M.; Likić, V.A.; Traven, A.; Lithgow, T. A small tim homohexamer in the relict mitochondrion of Cryptosporidium. Mol. Biol. Evol. 2011, 29, 113–122. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Guo, F.; Sun, M.; Zhu, G. A unique hexokinase in Cryptosporidium parvum, an apicomplexan pathogen lacking the Krebs cycle and oxidative phosphorylation. Protist 2014, 165, 701–714. [Google Scholar] [CrossRef]
- Stiff, R.E.; Davies, A.P.; Mason, B.W.; Hutchings, H.A.; Chalmers, R.M. Long-term health effects after resolution of acute Cryptosporidium parvum infection: A 1-year follow-up of outbreak-associated cases. J. Med. Microbiol. 2017, 66, 1607–1611. [Google Scholar] [CrossRef]
- Mammeri, M.; Chevillot, A.; Thomas, M.; Julien, C.; Auclair, E.; Pollet, T.; Polack, B.; Vallée, I.; Adjou, K.T. Cryptosporidium parvum-infected neonatal mice show gut microbiota remodelling using high-throughput sequencing analysis: Preliminary results. Acta Parasitol. 2019, 64, 268–275. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Funkhouser-Jones, L.J.; Akey, M.E.; Schaefer, D.A.; Ackman, K.; Riggs, M.W.; Stappenbeck, T.S.; Sibley, L.D. Neonatal Mouse Gut Metabolites Influence Cryptosporidium parvum Infection in Intestinal Epithelial Cells. mBio 2020, 11, e02520–e02582. [Google Scholar] [CrossRef]
- Cacciò, S.M.; Putignani, L. Epidemiology of Human Cryptosporidiosis. In Cryptosporidium: Parasite and Disease; Cacciò, S.M., Widmer, G., Eds.; Springer: Vienna, Austria, 2014; pp. 43–79. [Google Scholar]
- Bi, H.; Krausz, K.; Manna, S.; Li, F.; Johnson, C.; Gonzalez, F. Optimization of harvesting, extraction, and analytical protocols for UPLC-ESI-MS-based metabolomic analysis of adherent mammalian cancer cells. Anal. Bioanal. Chem. 2013, 405, 5279–5289. [Google Scholar] [CrossRef]
- Marcinowska, R.; Trygg, J.; Wolf-Watz, H.; Mortiz, T.; Surowiec, I. Optimization of a sample preparation method for the metabolomic analysis of clinically relevant bacteria. J. Microbiol. Meth. 2011, 87, 24–31. [Google Scholar] [CrossRef]
- Niklas, J.; Schneider, K.; Heinzle, E. Metabolic flux analysis in eukaryotes. Curr. Opin. Biotech. 2010, 21, 63–69. [Google Scholar] [CrossRef]
- Beale, D.J.; Morrison, P.D.; Karpe, A.V.; Dunn, M.S. Chemometric Analysis of Lavender Essential Oils Using Targeted and Untargeted GC-MS Acquired Data for the Rapid Identification and Characterization of Oil Quality. Molecules 2017, 22, 1339. [Google Scholar] [CrossRef] [PubMed]
- Beale, D.; Karpe, A.; Ahmed, W.; Cook, S.; Morrison, P.; Staley, C.; Sadowsky, M.; Palombo, E. A community multi-omics approach towards the assessment of surface water quality in an urban river system. Int. J. Environ. Res. Public Health 2017, 14, 303. [Google Scholar] [CrossRef] [PubMed]
- Kumarasingha, R.; Karpe, A.V.; Preston, S.; Yeo, T.-C.; Lim, D.S.L.; Tu, C.-L.; Luu, J.; Simpson, K.J.; Shaw, J.M.; Gasser, R.B.; et al. Metabolic profiling and in vitro assessment of anthelmintic fractions of Picria fel-terrae Lour. Int. J. Parasitol. Drugs Drug Resist. 2016, 6, 171–178. [Google Scholar] [CrossRef]
- Robinson, A.M.; Gondalia, S.V.; Karpe, A.V.; Eri, R.; Beale, D.J.; Morrison, P.D.; Palombo, E.A.; Nurgali, K. Fecal Microbiota and Metabolome in a Mouse Model of Spontaneous Chronic Colitis: Relevance to Human Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 2767–2787. [Google Scholar] [CrossRef]
- Beale, D.J.; Marney, D.; Marlow, D.R.; Morrison, P.D.; Dunn, M.S.; Key, C.; Palombo, E.A. Metabolomic analysis of Cryptosporidium parvum oocysts in water: A proof of concept demonstration. Environ. Pollut. 2013, 174, 201–203. [Google Scholar] [CrossRef]
- Koh, W.; Clode, P.L.; Monis, P.; Thompson, R.A. Multiplication of the waterborne pathogen Cryptosporidium parvum in an aquatic biofilm system. Parasites Vectors 2013, 6, 270. [Google Scholar] [CrossRef]
- Ng, J.S.Y.; Ryan, U.; Trengove, R.D.; Maker, G.L. Development of an untargeted metabolomics method for the analysis of human faecal samples using Cryptosporidium-infected samples. Mol. Biochem. Parasit. 2012, 185, 145–150. [Google Scholar] [CrossRef]
- Sarkar, S.; Hutton, M.L.; Vagenas, D.; Ruter, R.; Schüller, S.; Lyras, D.; Schembri, M.A.; Totsika, M. Intestinal colonization traits of pandemic multidrug-resistant Escherichia coli ST131. J. Infect. Dis. 2018, 218, 979–990. [Google Scholar] [CrossRef]
- Nielsen, K.L.; Dynesen, P.; Larsen, P.; Frimodt-Møller, N. Faecal Escherichia coli from patients with E. coli urinary tract infection and healthy controls who have never had a urinary tract infection. J. Med. Microbiol. 2014, 63, 582–589. [Google Scholar] [CrossRef]
- Ras, R.; Huynh, K.; Desoky, E.; Badawy, A.; Widmer, G. Perturbation of the intestinal microbiota of mice infected with Cryptosporidium parvum. Int. J. Parasitol. 2015, 45, 567–573. [Google Scholar] [CrossRef]
- Vemuri, R.; Gundamaraju, R.; Shinde, T.; Perera, A.P.; Basheer, W.; Southam, B.; Gondalia, S.V.; Karpe, A.V.; Beale, D.J.; Tristram, S. Lactobacillus acidophilus DDS-1 modulates intestinal-specific microbiota, short-chain fatty acid and immunological profiles in aging mice. Nutrients 2019, 11, 1297. [Google Scholar] [CrossRef] [PubMed]
- Rowin, J.; Xia, Y.; Jung, B.; Sun, J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol. Rep. 2017, 5, e13443. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.-H.; Rhee, M.-S.; Ahn, S.; Bang, B.-H.; Oh, J.E.; Lee, H.K.; Kim, B.-C. Faecalibaculum rodentium gen. nov., sp. nov., isolated from the faeces of a laboratory mouse. Antonie Van Leeuwenhoek 2015, 108, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, L.; Wang, Y.; Liu, C.; Zhang, L.; Zhu, S.; Liu, S.; Duan, L. Beneficial effect of butyrate-producing Lachnospiraceae on stress-induced visceral hypersensitivity in rats. J. Gastroenterol. Hepatol. 2019, 34, 1368–1376. [Google Scholar] [CrossRef]
- Park, J.-E.; Kim, J.-S.; Choi, S.-H.; Han, K.-I.; Kang, S.W.; Lee, J.H.; Oh, B.S.; Yu, S.Y.; Lee, K.C.; Eom, M.K.; et al. Draft genome sequence of Blautia sp. KGMB01111 isolated from a healthy Korean human faeces. Korean J. Microbiol. 2020, 56, 94–97. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wang, X.; Yu, X.; Hu, C.; Zhang, X. The family Coriobacteriaceae is a potential contributor to the beneficial effects of Roux-en-Y gastric bypass on type 2 diabetes. Surg. Obes. Relat. Dis. 2018, 14, 584–593. [Google Scholar] [CrossRef]
- Charania, R.; Wade, B.E.; McNair, N.N.; Mead, J.R. Changes in the Microbiome of Cryptosporidium-Infected Mice Correlate to Differences in Susceptibility and Infection Levels. Microorganisms 2020, 8, 879. [Google Scholar] [CrossRef] [PubMed]
- Mammeri, M.; Obregón, D.A.; Chevillot, A.; Polack, B.; Julien, C.; Pollet, T.; Cabezas-Cruz, A.; Adjou, K.T. Cryptosporidium parvum Infection Depletes Butyrate Producer Bacteria in Goat Kid Microbiome. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Kobayashi, J. d-Amino acids and lactic acid bacteria. Microorganisms 2019, 7, 690. [Google Scholar] [CrossRef]
- Shao, D.Y.; Bai, X.; Tong, M.W.; Zhang, Y.y.; Liu, X.l.; Zhou, Y.h.; Li, C.; Cai, W.; Gao, X.; Liu, M.; et al. Changes to the gut microbiota in mice induced by infection with Toxoplasma gondii. Acta Trop. 2020, 203, 105301. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kunisawa, A.; Hattori, T.; Kawana, S.; Kitada, Y.; Tamada, H.; Kawano, S.; Hayakawa, Y.; Iida, J.; Fukusaki, E. Free D-amino acids produced by commensal bacteria in the colonic lumen. Sci. Rep. 2018, 8, 17915. [Google Scholar] [CrossRef]
- Sasabe, J.; Miyoshi, Y.; Rakoff-Nahoum, S.; Zhang, T.; Mita, M.; Davis, B.M.; Hamase, K.; Waldor, M.K. Interplay between microbial d-amino acids and host d-amino acid oxidase modifies murine mucosal defence and gut microbiota. Nat. Microbiol. 2016, 1, 16125. [Google Scholar] [CrossRef]
- Xu, Z.; Guo, Y.; Roellig, D.M.; Feng, Y.; Xiao, L. Comparative analysis reveals conservation in genome organization among intestinal Cryptosporidium species and sequence divergence in potential secreted pathogenesis determinants among major human-infecting species. BMC Genomics 2019, 20, 406. [Google Scholar] [CrossRef]
- Rosa, L.T.; Bianconi, M.E.; Thomas, G.H.; Kelly, D.J. Tripartite ATP-independent periplasmic (TRAP) transporters and tripartite tricarboxylate transporters (TTT): From uptake to pathogenicity. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, R.M. Chapter Sixteen—Cryptosporidium. In Microbiology of Waterborne Diseases, 2nd ed.; Academic Press: London, UK, 2014; pp. 287–326. [Google Scholar]
- Shanmugasundram, A.; Gonzalez-Galarza, F.F.; Wastling, J.M.; Vasieva, O.; Jones, A.R. Library of apicomplexan metabolic pathways: A manually curated database for metabolic pathways of apicomplexan parasites. Nucleic Acids Res. 2012, 41, D706–D713. [Google Scholar] [CrossRef]
- Wang, R.; Wang, T.; Tait, S. Chapter 20—Metabolic regulation of immunity. In Kelley and Firestein’s Textbook of Rheumatology, 10th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 318–326. [Google Scholar]
- Gupta, S.; Igoillo-Esteve, M.; Michels, P.A.M.; Cordeiro, A.T. Glucose-6-Phosphate dehydrogenase of trypanosomatids: Characterization, target validation, and drug discovery. Mol. Biol. Int. 2011, 2011. [Google Scholar] [CrossRef]
- Choudhary, H.H.; Srivastava, P.N.; Singh, S.; Kumar, K.A.; Mishra, S. The shikimate pathway enzyme that generates chorismate is not required for the development of Plasmodium berghei in the mammalian host nor the mosquito vector. Int. J. Parasitol. 2018, 48, 203–209. [Google Scholar] [CrossRef]
- Rasmussen, K.R.; Arrowood, M.J.; Healey, M.C. Effectiveness of dehydroepiandrosterone in reduction of cryptosporidial activity in immunosuppressed rats. Antimicrob. Agents Chemother. 1992, 36, 220–222. [Google Scholar] [CrossRef]
- Chakraborty, S.; Roy, S.; Mistry, H.U.; Murthy, S.; George, N.; Bhandari, V.; Sharma, P. Potential Sabotage of Host Cell Physiology by Apicomplexan Parasites for Their Survival Benefits. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Yu, X.; Guo, F.; Mouneimne, R.B.; Zhu, G. Cryptosporidium parvum Elongation Factor 1α Participates in the Formation of Base Structure at the Infection Site During Invasion. J. Infect. Dis. 2019, 221, 1816–1825. [Google Scholar] [CrossRef]
- Elliott, D.A.; Clark, D.P. Cryptosporidium parvum induces host cell actin accumulation at the host-parasite interface. Infect. Immun. 2000, 68, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Matson, E.; Reginato, A.M. Chapter 23—Crystalline disorders associated with renal disease including oxalate arthropathy. In Gout & Other Crystal Arthropathies; Terkeltaub, R., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2012; pp. 282–292. [Google Scholar]
- Engelking, L.R. Chapter 27—Metabolic fates of pyruvate. In Textbook of Veterinary Physiological Chemistry, 3rd ed.; Engelking, L.R., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 169–173. [Google Scholar]
- Lai, C.; Pursell, N.; Gierut, J.; Saxena, U.; Zhou, W.; Dills, M.; Diwanji, R.; Dutta, C.; Koser, M.; Nazef, N.; et al. Specific inhibition of hepatic lactate dehydrogenase reduces oxalate production in mouse models of primary hyperoxaluria. Mol. Ther. 2018, 26, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Wirth, J.D.; Boucher, J.I.; Jacobowitz, J.R.; Classen, S.; Theobald, D.L. Functional and structural resilience of the active site loop in the evolution of Plasmodium lactate dehydrogenase. Biochemistry 2018, 57, 6434–6442. [Google Scholar] [CrossRef]
- Mokhbatly, A.; Selim, A. Clinicopathological studies on Cryptosporidium infection in newly born calves. Vet. Med. J. 2003, 1, 101–110. [Google Scholar]
- Htun, N.S.N.; Odermatt, P.; Müller, I.; Yap, P.; Steinmann, P.; Schindler, C.; Gerber, M.; Du Randt, R.; Walter, C.; Pühse, U.; et al. Association between gastrointestinal tract infections and glycated hemoglobin in school children of poor neighborhoods in Port Elizabeth, South Africa. PLOS Neglected Trop. Dis. 2018, 12, e0006332. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Li, H. Gut Microbiota and Iron: The Crucial Actors in Health and Disease. Pharmaceuticals 2018, 11, 98. [Google Scholar] [CrossRef]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Griffin, J.L.; Nicholls, A.W.; Daykin, C.A.; Heald, S.; Keun, H.C.; Schuppe-Koistinen, I.; Griffiths, J.R.; Cheng, L.L.; Rocca-Serra, P.; Rubtsov, D.V.; et al. Standard reporting requirements for biological samples in metabolomics experiments: Mammalian/in vivo experiments. Metabolomics 2007, 3, 179–188. [Google Scholar] [CrossRef]
- UK-Environmental-Agency. The Microbiology of Drinking Water (2010)—Part 14—Methods for the Isolation, Identification and Enumeration of Cryptosporidium Oocysts and Giardia Cysts. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/755552/Part_14-oct20-234.pdf (accessed on 21 May 2021).
- Wilson, N.A.; Park, H.S.; Lee, K.S.; Barron, L.K.; Warner, B.W. A Novel Approach to Calculating Small Intestine Length Based on Magnetic Resonance Enterography. J. Am. Coll. Surg. 2017, 225, 266–273.e261. [Google Scholar] [CrossRef]
- Kirman, C.R.; Hays, S.M.; Aylward, L.L.; Suh, M.; Harris, M.A.; Thompson, C.M.; Haws, L.C.; Proctor, D.M. Physiologically based pharmacokinetic model for rats and mice orally exposed to chromium. Chem. Biol. Interact. 2012, 200, 45–64. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics by gas chromatography–mass spectrometry: Combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30–34. [Google Scholar] [CrossRef]
- Yuan, B.-F.; Zhu, Q.-F.; Guo, N.; Zheng, S.-J.; Wang, Y.-L.; Wang, J.; Xu, J.; Liu, S.-J.; He, K.; Hu, T.; et al. Comprehensive Profiling of Fecal Metabolome of Mice by Integrated Chemical Isotope Labeling-Mass Spectrometry Analysis. Anal. Chem. 2018, 90, 3512–3520. [Google Scholar] [CrossRef]
- O’Sullivan, V.; Madrid-Gambin, F.; Alegra, T.; Gibbons, H.; Brennan, L. Impact of Sample Storage on the NMR Fecal Water Metabolome. ACS Omega 2018, 3, 16585–16590. [Google Scholar] [CrossRef]
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24. [Google Scholar] [CrossRef]
- Beale, D.J.; Oh, D.Y.; Karpe, A.V.; Tai, C.; Dunn, M.S.; Tilmanis, D.; Palombo, E.A.; Hurt, A.C. Untargeted metabolomics analysis of the upper respiratory tract of ferrets following influenza A virus infection and oseltamivir treatment. Metabolomics 2019, 15, 33. [Google Scholar] [CrossRef] [PubMed]
- Beale, D.; Karpe, A.; Rupprecht, U. In-Time TMS Derivatization and GC/MS Determination of Sugars, Organic Acids and Amino Acids for High Throughput Metabolomics Studies; KG: Mülheim an der Ruhr, Germany, 2020; pp. 1–10. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Arndt, D.; Xia, J.; Liu, Y.; Zhou, Y.; Guo, A.C.; Cruz, J.A.; Sinelnikov, I.; Budwill, K.; Nesbø, C.L.; Wishart, D.S. Metagenassist: A comprehensive web server for comparative metagenomics. Nucleic Acids Res. 2012, 40, W88–W95. Available online: http://nar.oxfordjournals.org/content/40/W1/W88.abstract (accessed on 21 May 2021). [CrossRef]
- Hernández-de-Diego, R.; Tarazona, S.; Martínez-Mira, C.; Balzano-Nogueira, L.; Furió-Tarí, P.; Pappas, G.J., Jr.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–w509. [Google Scholar] [CrossRef]






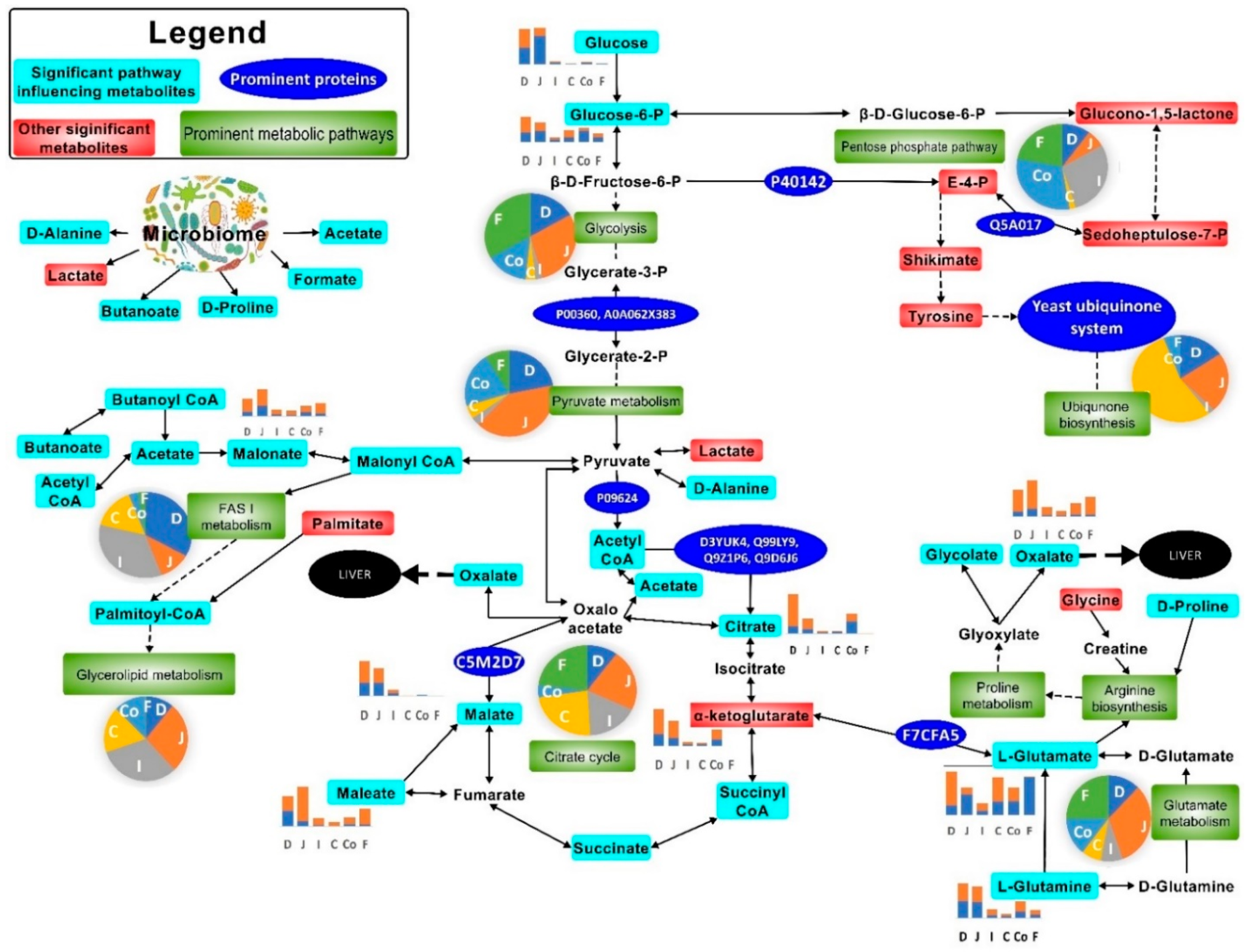
 ) and Cryptosporidium (
) and Cryptosporidium ( ) infected mice. Note: Please refer to Figure S8 for a more descriptive pathway chart.
) and Cryptosporidium () infected mice. Note: Please refer to Figure S8 for a more descriptive pathway chart.
) infected mice. Note: Please refer to Figure S8 for a more descriptive pathway chart.
) and Cryptosporidium () infected mice. Note: Please refer to Figure S8 for a more descriptive pathway chart.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Match Status | Impact | FDR |
|---|---|---|---|
| Arginine biosynthesis | 13/27 | 1.12 | <0.0001 |
| Citrate cycle (TCA cycle) | 15/42 | 1.95 | <0.0001 |
| Glycolysis or Gluconeogenesis | 16/61 | 1.28 | <0.0001 |
| Pyruvate metabolism | 13/45 | 0.93 | <0.0001 |
| Nitrogen metabolism | 6/10 | 1.00 | 0.0002 |
| Glutathione metabolism | 13/56 | 0.69 | 0.0006 |
| Alanine, aspartate, and glutamate metabolism | 13/61 | 0.83 | 0.0015 |
| Glyoxylate and dicarboxylate metabolism | 12/56 | 0.53 | 0.0023 |
| Galactose metabolism | 11/51 | 0.66 | 0.0035 |
| Arginine and proline metabolism | 14/78 | 0.52 | 0.0039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpe, A.V.; Hutton, M.L.; Mileto, S.J.; James, M.L.; Evans, C.; Shah, R.M.; Ghodke, A.B.; Hillyer, K.E.; Metcalfe, S.S.; Liu, J.-W.; et al. Cryptosporidiosis Modulates the Gut Microbiome and Metabolism in a Murine Infection Model. Metabolites 2021, 11, 380. https://doi.org/10.3390/metabo11060380
Karpe AV, Hutton ML, Mileto SJ, James ML, Evans C, Shah RM, Ghodke AB, Hillyer KE, Metcalfe SS, Liu J-W, et al. Cryptosporidiosis Modulates the Gut Microbiome and Metabolism in a Murine Infection Model. Metabolites. 2021; 11(6):380. https://doi.org/10.3390/metabo11060380
Chicago/Turabian StyleKarpe, Avinash V., Melanie L. Hutton, Steven J. Mileto, Meagan L. James, Chris Evans, Rohan M. Shah, Amol B. Ghodke, Katie E. Hillyer, Suzanne S. Metcalfe, Jian-Wei Liu, and et al. 2021. "Cryptosporidiosis Modulates the Gut Microbiome and Metabolism in a Murine Infection Model" Metabolites 11, no. 6: 380. https://doi.org/10.3390/metabo11060380
APA StyleKarpe, A. V., Hutton, M. L., Mileto, S. J., James, M. L., Evans, C., Shah, R. M., Ghodke, A. B., Hillyer, K. E., Metcalfe, S. S., Liu, J.-W., Walsh, T., Lyras, D., Palombo, E. A., & Beale, D. J. (2021). Cryptosporidiosis Modulates the Gut Microbiome and Metabolism in a Murine Infection Model. Metabolites, 11(6), 380. https://doi.org/10.3390/metabo11060380