
Disparate Metabolomic Responses to Fructose Consumption between Different Mouse Strains and the Role of Gut Microbiota




Abstract
1. Introduction
2. Results
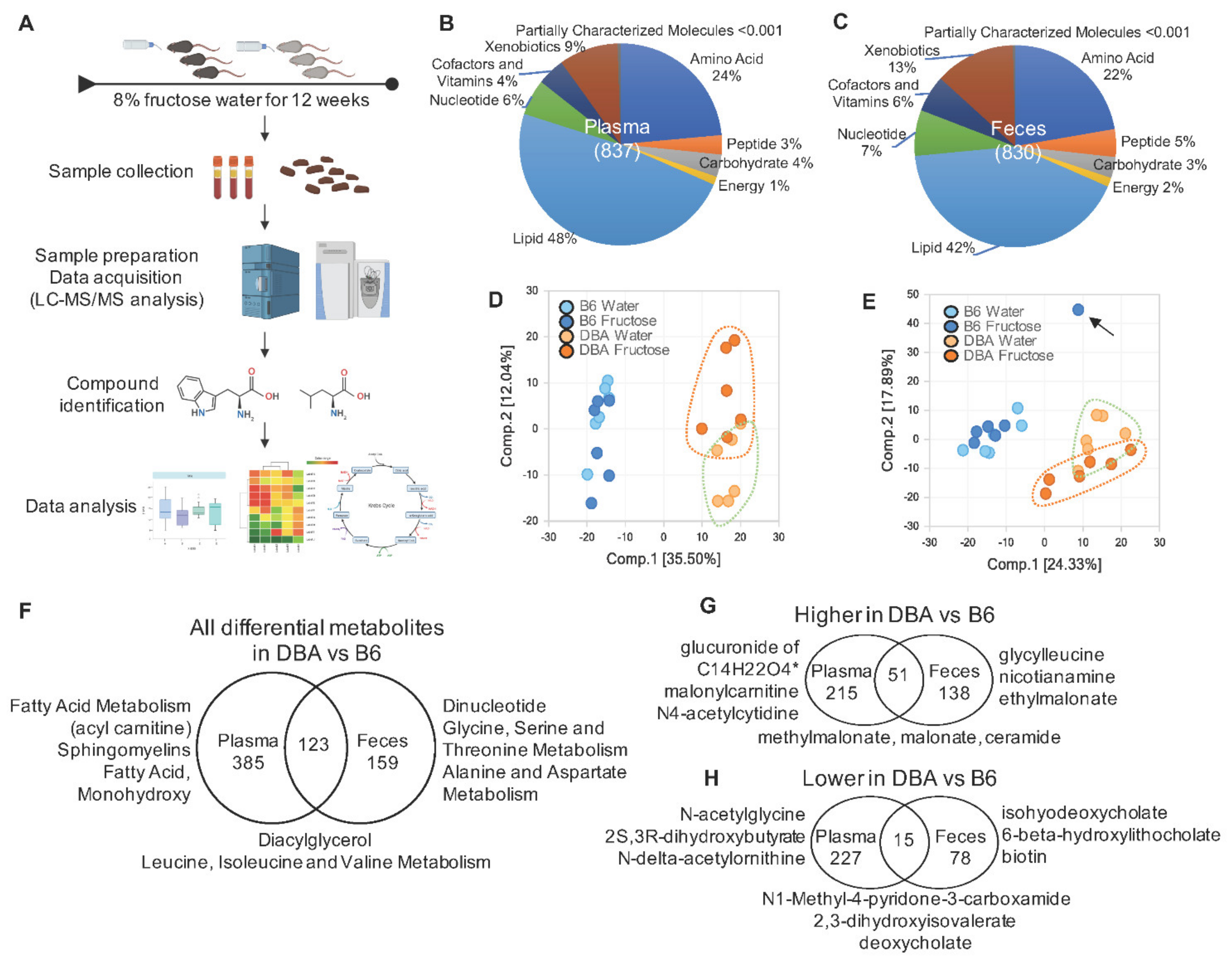
2.1. Disparate Metabolomic Profiles between DBA and B6 Mice
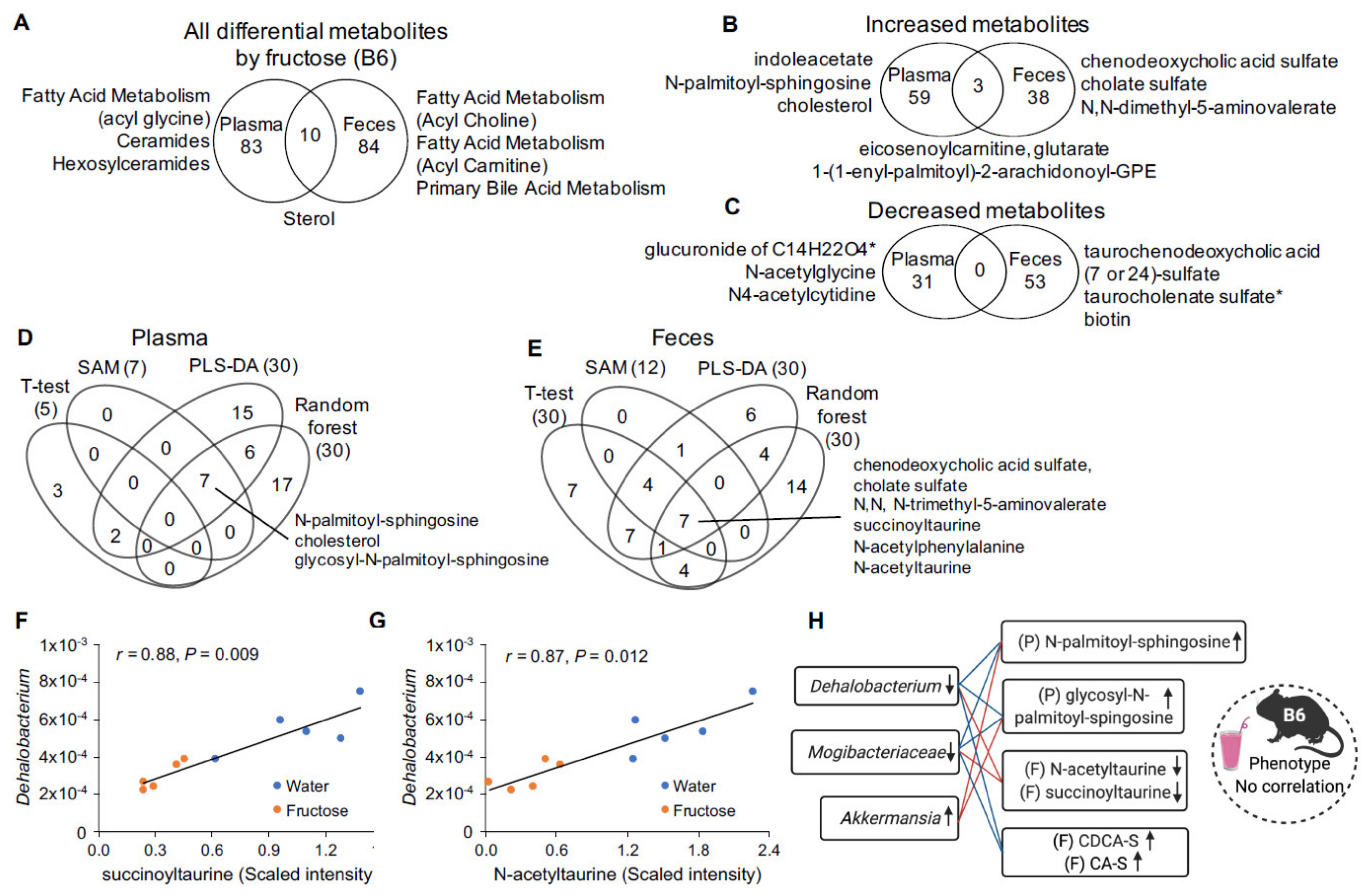
2.2. Fructose-Responsive Metabolites in DBA Mice
2.3. Correlation of Fructose-Responsive Metabolites with Metabolic Phenotypes or Fecal Microbiota in DBA Mice
2.4. Fructose-Responsive Metabolites in B6 Mice
2.5. Correlation of Fructose-Responsive Metabolites with Fecal Microbiota and Metabolic Phenotypes in B6 Mice
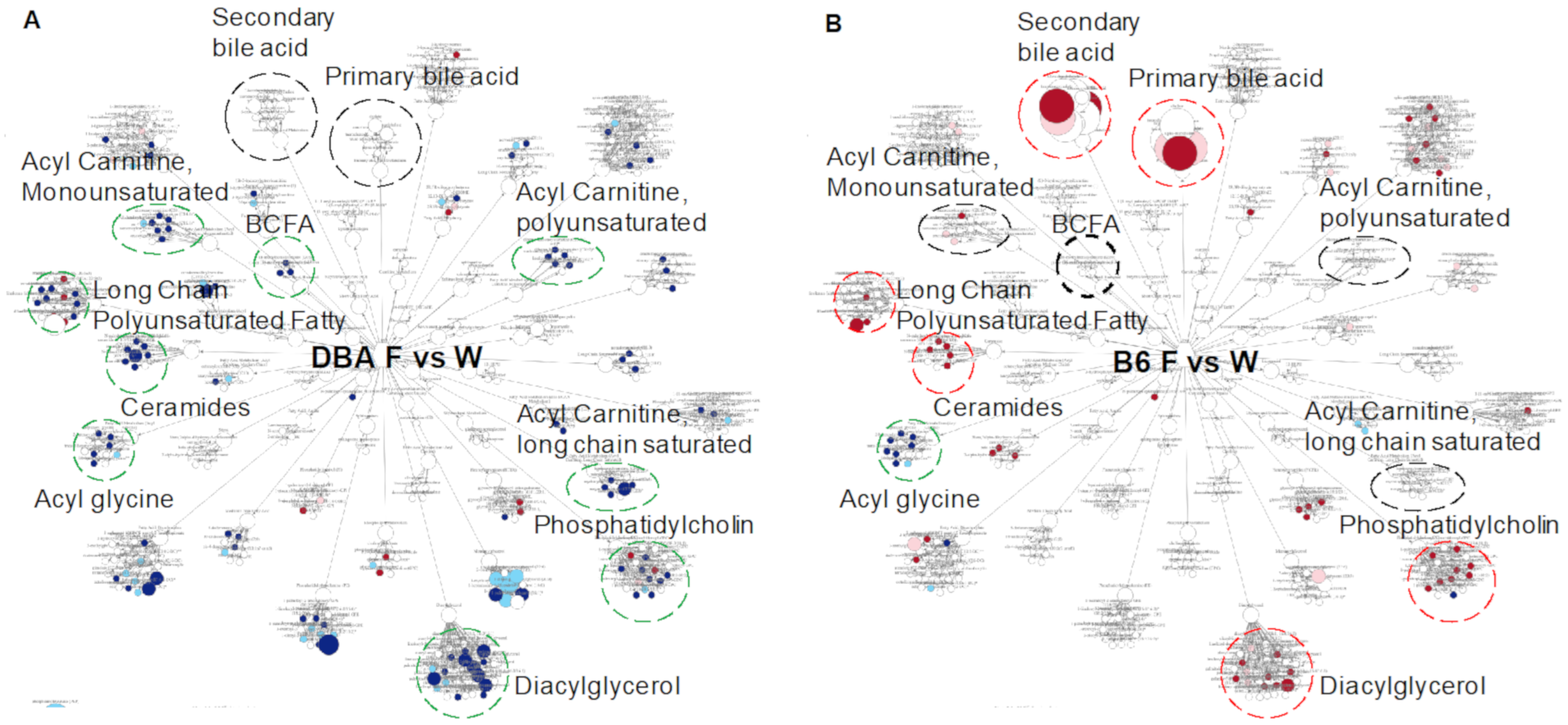
2.6. Comparison between DBA and B6 Reveals Strain-Specific Metabolic Pathways in Response to Fructose Consumption
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Sample Preparation and LC-MS/MS Analysis
4.3. Data Extraction and Metabolites Identification
4.4. Metabolites Quantification and Data Normalization
4.5. Principal Component Analysis and Differential Metabolites
4.6. Metabolic Pathway Analysis of Significantly Altered Metabolites
4.7. Prioritization of Statistically Robust Differential Metabolites
4.8. Correlation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marriott, B.P.; Cole, N.; Lee, E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009, 139, 1228S–1235S. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, D.; Korem, T.; Zmora, N.; Israeli, D.; Rothschild, D.; Weinberger, A.; Ben-Yacov, O.; Lador, D.; Avnit-Sagi, T.; Lotan-Pompan, M.; et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell 2015, 163, 1079–1095. [Google Scholar] [CrossRef]
- Yang, X. Multitissue multiomics systems biology to dissect complex diseases. Trends Mol. Med. 2020, 26, 718–728. [Google Scholar] [CrossRef]
- Glendinnning, J.; Breinager, L.; Kyrillou, E.; Lacuna, K.; Rocha, R.; Sclafani, A. Differential effects of sucrose and fructose on dietary obesity in four mouse strains. Physiol. Behav. 2010, 101, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Fiveash, C.E.; Braude, J.P.; Osborne, B.; Brown, S.H.J.; Mitchell, T.W.; Turner, N. Disparate metabolic response to fructose feeding between different mouse strains. Sci. Rep. 2015, 5, 18474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Byun, H.R.; Ying, Z.; Blencowe, M.; Zhao, Y.; Hong, J.; Shu, L.; Chella, K.; Gomez-Pinilla, F.; Yang, X. Differential metabolic and multi-tissue transcriptomic responses to fructose consumption among genetically diverse mice. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165569. [Google Scholar] [CrossRef]
- Ahn, I.S.; Lang, J.M.; Olson, C.A.; Diamante, G.; Zhang, G.; Ying, Z.; Byun, H.R.; Cely, I.; Ding, J.; Cohn, P.; et al. Host Genetic Background and Gut Microbiota Contribute to Differential Metabolic Responses to High Fructose Consumption in Mice. J. Nutr. 2020, 150, 2716–2728. [Google Scholar] [CrossRef] [PubMed]
- Goff, L.M.; Whyte, M.B.; Samuel, M.; Harding, S.V. Significantly greater triglyceridemia in Black African compared to White European men following high added fructose and glucose feeding: A randomized crossover trial. Lipids Health Dis. 2016, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Panda, C.; Voruganti, V.S. Heterogeneity in metabolic responses to dietary fructose. Front. Genet. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bar, N.; Korem, T.; Weissbrod, O.; Zeevi, D.; Rothschild, D.; Leviatan, S.; Kosower, N.; Lotan-Pompan, M.; Weinberger, A.; Le Roy, C.I.; et al. A reference map of potential determinants for the human serum metabolome. Nature 2020, 588, 135–140. [Google Scholar] [CrossRef]
- Newgard, C.B. Review Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 2017, 25, 43–56. [Google Scholar] [CrossRef]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Su, X.; Magkos, F.; Zhou, D.; Eagon, J.C.; Fabbrini, E.; Okunade, A.L.; Klein, S. Adipose tissue monomethyl branched-chain fatty acids and insulin sensitivity: Effects of obesity and weight loss. Obesity 2015, 23, 329–334. [Google Scholar] [CrossRef]
- Neis, E.P.J.G.; Dejong, C.H.C.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A BCAA Related Metabolic Signature that differentiates obese and lean. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Pakiet, A.; Wilczynski, M.; Rostkowska, O.; Korczynska, J.; Jabłonska, P.; Kaska, L.; Proczko-Stepaniak, M.; Sobczak, E.; Stepnowski, P.; Magkos, F.; et al. The Effect of One Anastomosis Gastric Bypass on Branched-Chain Fatty Acid and Branched-Chain Amino Acid Metabolism in Subjects with Morbid Obesity. Obes. Surg. 2020, 30, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829–1840. [Google Scholar] [CrossRef]
- Li, Z.; Quan, G.; Jiang, X.; Yang, Y.; Ding, X.; Zhang, D.; Wang, X.; Hardwidge, P.R.; Ren, W.; Zhu, G. Effects of metabolites derived from gut microbiota and hosts on pathogens. Front. Cell. Infect. Microbiol. 2018, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D.; Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; et al. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361. [Google Scholar] [CrossRef]
- Herman, M.A.; Samuel, V.T. The sweet path to metabolic demise: Fructose and lipid synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef]
- Gonzalez-Granda, A.; Damms-Machado, A.; Basrai, M.; Bischoff, S.C. Changes in plasma acylcarnitine and lysophosphatidylcholine levels following a high-fructose diet: A targeted metabolomics study in healthy women. Nutrients 2018, 10, 1254. [Google Scholar] [CrossRef]
- Liu, M.; Chu, J.; Gu, Y.; Shi, H.; Zhang, R.; Wang, L.; Chen, J.; Shen, L.; Yu, P.; Chen, X.; et al. Serum N1-methylnicotinamide is associated with coronary artery disease in Chinese patients. J. Am. Heart Assoc. 2017, 6, e004328. [Google Scholar] [CrossRef]
- Liu, M.; Li, L.; Chu, J.; Zhu, B.; Zhang, Q.; Yin, X.; Jiang, W.; Dai, G.; Ju, W.; Wang, Z.; et al. Serum N1-methylnicotinamide is associated with obesity and diabetes in Chinese. J. Clin. Endocrinol. Metab. 2015, 100, 3112–3117. [Google Scholar] [CrossRef]
- Romano, A.; Coccurello, R.; Giacovazzo, G.; Bedse, G.; Moles, A.; Gaetani, S. Oleoylethanolamide: A novel potential pharmacological alternative to cannabinoid antagonists for the control of appetite. BioMed Res. Int. 2014, 2014, 203425. [Google Scholar] [CrossRef]
- Zhao, M.; Zhao, L.; Xiong, X.; He, Y.; Huang, W.; Liu, Z.; Ji, L.; Pan, B.; Guo, X.; Wang, L.; et al. TMAVA, a Metabolite of Intestinal Microbes, Is Increased in Plasma from Patients with Liver Steatosis, Inhibits γ-Butyrobetaine Hydroxylase, and Exacerbates Fatty Liver in Mice. Gastroenterology 2020, 158, 2266–2281. [Google Scholar] [CrossRef]
- Wahlstrm, A.; Sayin, S.I.; Marschall, H.U.; Buckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Cariello, M.; Piccinin, E.; Garcia-Irigoyen, O.; Sabbà, C.; Moschetta, A. Nuclear receptor FXR, bile acids and liver damage: Introducing the progressive familial intrahepatic cholestasis with FXR mutations. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Fujisaka, S.; Avila-Pacheco, J.; Soto, M.; Kostic, A.; Dreyfuss, J.M.; Pan, H.; Ussar, S.; Altindis, E.; Li, N.; Bry, L.; et al. Diet, Genetics, and the Gut Microbiome Drive Dynamic Changes in Plasma Metabolites. Cell Rep. 2018, 22, 3072–3086. [Google Scholar] [CrossRef]
- Stöckli, J.; Fisher-Wellman, K.H.; Chaudhuri, R.; Zeng, X.Y.; Fazakerley, D.J.; Meoli, C.C.; Thomas, K.C.; Hoffman, N.J.; Mangiafico, S.P.; Xirouchaki, C.E.; et al. Metabolomic analysis of insulin resistance across different mouse strains and diets. J. Biol. Chem. 2017, 292, 19135–19145. [Google Scholar] [CrossRef]
- Silva, J.C.P.; Mota, M.; Martins, F.O.; Nogueira, C.; Gonçalves, T.; Carneiro, T.; Pinto, J.; Duarte, D.; Barros, A.S.; Jones, J.G.; et al. Intestinal Microbial and Metabolic Profiling of Mice Fed with High-Glucose and High-Fructose Diets. J. Proteome Res. 2018, 17, 2880–2891. [Google Scholar] [CrossRef] [PubMed]
- Meissen, J.K.; Hirahatake, K.M.; Adams, S.H.; Fiehn, O. Temporal metabolomic responses of cultured HepG2 liver cells to high fructose and high glucose exposures John. Physiol. Behav. 2015, 11, 707–721. [Google Scholar]
- Topping, D.L.; Mayes, P.A. The immediate effects of insulin and fructose on the metabolism of the perfused liver. Changes in lipoprotein secretion, fatty acid oxidation and esterification, lipogenesis and carbohydrate metabolism. Biochem. J. 1972, 126, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Sayed, A.; Binnert, C.; Lê, K.A.; Bortolotti, M.; Schneiter, P.; Tappy, L. A high-fructose diet impairs basal and stress-mediated lipid metabolism in healthy male subjects. Br. J. Nutr. 2008, 100, 393–399. [Google Scholar] [CrossRef]
- Kaneda, T. Iso- and anteiso-fatty acids in bacteria: Biosynthesis, function, and taxonomic significance. Microbiol. Rev. 1991, 55, 288–302. [Google Scholar] [CrossRef]
- Gebicki, J.; Sysa-Jedrzejowska, A.; Adamus, J.; Woźniacka, A.; Rybak, M.; Zielonka, J. 1-Methylnicotinamide: A potent anti-inflammatory agent of vitamin origin. Pol. J. Pharmacol. 2003, 55, 109–112. [Google Scholar]
- Liu, F.; Smith, A.D.; Solano-Aguilar, G.; Wang, T.T.Y.; Pham, Q.; Beshah, E.; Tang, Q.; Urban, J.F.; Xue, C.; Li, R.W. Mechanistic insights into the attenuation of intestinal inflammation and modulation of the gut microbiome by krill oil using in vitro and in vivo models. Microbiome 2020, 8, 1–21. [Google Scholar] [CrossRef]
- Libert, D.M.; Nowacki, A.S.; Natowicz, M.R. Metabolomic analysis of obesity, metabolic syndrome, and type 2 diabetes: Amino acid and acylcarnitine levels change along a spectrum of metabolic wellness. PeerJ 2018, 6, e5410. [Google Scholar] [CrossRef]
- Kuryłowicz, A.; Cakała-Jakimowicz, M.; Puzianowska-Kuźnicka, M. Targeting abdominal obesity and its complications with dietary phytoestrogens. Nutrients 2020, 12, 582. [Google Scholar] [CrossRef]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Neale, G.; Lewis, B.; Weaver, V.; Panveliwalla, D. Serum bile acids in liver disease. Gut 1971, 12, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and Sugar: A major mediator of nonalcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.R.F.; Pattni, S.S. Managing bile acid diarrhoea. Ther. Adv. Gastroenterol. 2010, 3, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.; Azuma, J.; Mozaffari, M. Role of antioxidant activity of taurine in diabetes. Can. J. Physiol. Pharmacol. 2009, 87, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.; Kim, H.W. Effects and mechanisms of taurine as a therapeutic agent. Biomol. Ther. 2018, 26, 225–241. [Google Scholar] [CrossRef]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef]
- Evans, A.M.; Bridgewater, B.R.; Liu, Q.; Mitchell, M.; Robinson, R.J.; Dai, H.; Stewart, S.J.; DeHaven, C.D.; Miller, L. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high-throughput profiling metabolomics. Metabolomics 2014, 4, 1000132. [Google Scholar]
- Ford, L.; Kennedy, A.D.; Goodman, K.D.; Pappan, K.L.; Evans, A.M.; Miller, L.A.D.; Wulff, J.E.; Wiggs, B.R.; Lennon, J.J.; Elsea, S.; et al. Precision of a clinical metabolomics profiling platform for use in the identification of inborn errors of metabolism. J. Appl. Lab. Med. 2020, 5, 342–356. [Google Scholar] [CrossRef]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steve, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 70–795. [Google Scholar] [CrossRef]
- Loftfield, E.; Vogtmann, E.; Sampson, J.N.; Moore, S.C.; Nelson, H.; Knight, R.; Chia, N.; Sinha, R. Comparison of collection methods for fecal samples for discovery metabolomics in epidemiologic studies. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1483–1890. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.H.; Yuan, L.; Sha, W.; Sun, Z.L. Gene differential coexpression analysis based on biweight correlation and maximum clique. BMC Bioinform. 2014, 15, S3. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Change 2 | Rikenellaceae (Unknown) | Pseudomonadaceae (Unknown) | Pseudomonas | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Metabolite | Sample 1 | Pathway | Subpathway | Corr 3 | FDR 4 | Corr | FDR | Corr | FDR | |
| 1,5-anhydroglucitol (1,5-AG) | Plasma | Carbohydrate | Glycolysis, Gluconeogenesis, and Pyruvate Metabolism | 1.32 * | −0.91 | 0.007 | ||||
| 1-oleoyl-2-arachidonoyl-GPI (18:1/20:4) # | Plasma | Lipid | Phosphatidylinositol (Pl) | 1.45 * | 0.73 | 0.034 | ||||
| 1-methyl-5-imidazoleacetate | Feces | Amino Acid | Histidine Metabolism | 2.03 * | −0.68 | 0.037 | 0.52 | 0.099 | 0.72 | 0.035 |
| 4-hydroxyphenylacetate | Feces | Amino Acid | Phenylalanine Metabolism | 2.84 ** | −0.64 | 0.046 | 0.70 | 0.035 | 0.71 | 0.035 |
| protoporphyrin IX | Feces | Cofactors and Vitamins | Hemoglobin and Porphyrin Metabolism | 0.24 ** | 0.64 | 0.045 | −0.64 | 0.045 | ||
| behenoyl ethanolamide (22:0) # | Feces | Lipid | Endocannabinoid | 0.49 * | 0.79 | 0.019 | −0.67 | 0.039 | −0.65 | 0.045 |
| lignoceroyl ethanolamide (24:0) # | Feces | Lipid | Endocannabinoid | 0.50 * | 0.73 | 0.035 | −0.68 | 0.037 | −0.59 | 0.063 |
| 2-hydroxyarachidate # | Feces | Lipid | Fatty Acid, Monohydroxy | 0.34 ** | 0.63 | 0.045 | −0.71 | 0.035 | −0.70 | 0.035 |
| 2-hydroxybehenate | Feces | Lipid | Fatty Acid, Monohydroxy | 0.29 ** | 0.67 | 0.039 | −0.71 | 0.035 | −0.70 | 0.035 |
| 2-hydroxylignocerate # | Feces | Lipid | Fatty Acid, Monohydroxy | 0.41 ** | 0.64 | 0.045 | −0.85 | 0.010 | −0.69 | 0.035 |
| 2-hydroxynervonate # | Feces | Lipid | Fatty Acid, Monohydroxy | 0.46 ** | 0.72 | 0.035 | −0.76 | 0.026 | −0.64 | 0.045 |
| choline | Feces | Lipid | Phospholipid Metabolism | 1.54 * | −0.89 | 0.006 | ||||
| taurohyodeoxycholic acid | Feces | Lipid | Secondary Bile Acid Metabolism | 2.20 ** | 0.80 | 0.02 | ||||
| cholesterol | Feces | Lipid | Sterol | 0.67 * | 0.78 | 0.019 | −0.75 | 0.028 | ||
| sphinganine | Feces | Lipid | Sphingolipid Synthesis | 0.53 * | 0.69 | 0.036 | −0.68 | 0.037 | −0.63 | 0.045 |
| lysylleucine | Feces | Peptide | Dipeptide | 3.24 * | 0.86 | 0.010 | 0.69 | 0.035 | ||
| apigenin | Feces | Xenobiotics | Food Component/Plant | 2.22 * | −0.78 | 0.019 | ||||
| coumestrol | Feces | Xenobiotics | Food Component/Plant | 3.00 ** | −0.78 | 0.020 | ||||
| enterodiol | Feces | Xenobiotics | Food Component/Plant | 2.06 * | −0.84 | 0.011 | 0.62 | 0.048 | ||
| enterolactone | Feces | Xenobiotics | Food Component/Plant | 4.04 ** | −0.66 | 0.039 | ||||
| pinosylvin | Feces | Xenobiotics | Food Component/Plant | 6.36 * | −0.66 | 0.039 | ||||
| Fold Change 2 | Dehalobacterium | Magibacteriaceae (Unknown) | Akkermansia | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Metabolite | Sample 1 | Pathway | Subpathway | Corr 3 | FDR 4 | Corr | FDR | Corr | FDR | |
| Indoleacetate | Plasma | Amino Acid | Tryptophan Metabolism | 1.78 ** | −0.83 | 0.004 | −0.85 | 0.003 | 0.64 | 0.041 |
| Carotene diol | Plasma | Cofactors/Vitamins | Vitamin A Metabolism | 1.69 * | −0.79 | 0.008 | −0.77 | 0.010 | 0.88 | 0.002 |
| Glycosyl-N-palmitoyl-sphingosine (d18:1/16:0) | Plasma | Lipid | Hexosylceramides | 1.42 ** | −0.77 | 0.010 | −0.76 | 0.010 | 0.83 | 0.004 |
| Cholesterol | Plasma | Lipid | Sterol | 1.30 * | −0.63 | 0.041 | −0.64 | 0.041 | 0.64 | 0.041 |
| N-palmitoyl-sphingosine (d18:1/16:0) | Plasma | Lipid | Ceramides | 1.63 ** | −0.76 | 0.001 | −0.85 | 0.003 | 0.73 | 0.014 |
| Sphingomyelin (d18:1/14:0, d16:1/16:0) | Plasma | Lipid | Sphingomyelins | 1.44 * | −0.89 | 0.002 | −0.84 | 0.003 | 0.94 | 0.001 |
| Sphingomyelin (d17:1/16:0, d18:1/15:0, d16:1/17:0) | Plasma | Lipid | Sphingomyelins | 1.38 * | −0.88 | 0.002 | -0.78 | 0.010 | 0.90 | 0.002 |
| Fold Change 2 | Dehalobacterium | Magibacteriaceae (Unknown) | Akkermansia | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Metabolite | Sample 1 | Pathway | Subpathway | Corr 3 | FDR 4 | Corr | Adjp | Corr | Adjp | |
| Isobutyrylglycine (C4) | Feces | Amino Acid | Leucine, Isoleucine and Valine | 3.32 * | −0.77 | 0.033 | ||||
| N,N-dimethyl-5-aminovalerate | Feces | Amino Acid | Lysine Metabolism | 1.54 * | −0.75 | 0.038 | 0.77 | 0.036 | ||
| N-acetyltaurine | Feces | Amino Acid | Methionine, Cysteine, SAM and Taurine Metabolism | 0.21 * | 0.87 | 0.013 | 0.89 | 0.008 | ||
| Succinoyltaurine | Feces | Amino Acid | Methionine, Cysteine, SAM and Taurine Metabolism | 0.31 * | 0.88 | 0.009 | 0.94 | 0.002 | ||
| Taurine | Feces | Amino Acid | Methionine, Cysteine, SAM and Taurine Metabolism | 0.34 * | 0.85 | 0.017 | −0.80 | 0.026 | ||
| Biotin | Feces | Cofactors/ Vitamins | Biotin Metabolism | 0.19 ** | 0.81 | 0.025 | ||||
| Phenylacetyltaurine | Feces | Lipid | Acetylated Peptides | 0.40 * | 0.79 | 0.027 | 0.92 | 0.004 | −0.78 | 0.032 |
| N-linoleoyltaurine | Feces | Lipid | Endocannabinoid | 0.34 * | −0.73 | 0.045 | ||||
| Stearoyl ethanolamide | Feces | Lipid | Endocannabinoid | 2.07 * | −0.74 | 0.045 | −0.83 | 0.022 | ||
| Lactosyl-N-palmitoyl-sphingosine (d18:1/16:0) | Feces | Lipid | Lactosylceramides | 0.45 * | 0.95 | 0.002 | 0.79 | 0.027 | ||
| Chenodeoxycholic acid- sulfate | Feces | Lipid | Primary Bile Acid | 8.61 ** | −0.80 | 0.025 | −0.74 | 0.045 | ||
| Cholate Sulfate | Feces | Lipid | Primary Bile Acid | 8.45 ** | −0.84 | 0.017 | −0.75 | 0.038 | ||
| Tauro-beta muricholate | Feces | Lipid | Primary Bile Acid | 0.11 ** | 0.88 | 0.010 | 0.81 | 0.025 | ||
| Taurocholate | Feces | Lipid | Primary Bile Acid | 0.34 * | 0.77 | 0.036 | 0.76 | 0.036 | ||
| Taurochenodeoxycholic acid (7 or 24)-sulfate | Feces | Lipid | Secondary Bile Acid | 0.00 ** | −0.78 | 0.032 | ||||
| Taurohyodeoxycholic acid | Feces | Lipid | Secondary Bile Acid | 0.16 ** | 0.82 | 0.024 | −0.82 | 0.025 | ||
| Beta-sitosterol | Feces | Lipid | Sterol | 0.66 * | 0.91 | 0.004 | 0.81 | 0.025 | ||
| Campesterol | Feces | Lipid | Sterol | 0.64 * | 0.92 | 0.004 | 0.82 | 0.025 | ||
| Cholesterol | Feces | Lipid | Sterol | 0.70 * | 0.76 | 0.037 | −0.76 | 0.036 | ||
| Stigmasterol | Feces | Lipid | Sterol | 0.66 * | 0.92 | 0.004 | 0.85 | 0.017 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, I.-S.; Yoon, J.; Diamante, G.; Cohn, P.; Jang, C.; Yang, X. Disparate Metabolomic Responses to Fructose Consumption between Different Mouse Strains and the Role of Gut Microbiota. Metabolites 2021, 11, 342. https://doi.org/10.3390/metabo11060342
Ahn I-S, Yoon J, Diamante G, Cohn P, Jang C, Yang X. Disparate Metabolomic Responses to Fructose Consumption between Different Mouse Strains and the Role of Gut Microbiota. Metabolites. 2021; 11(6):342. https://doi.org/10.3390/metabo11060342
Chicago/Turabian StyleAhn, In-Sook, Justin Yoon, Graciel Diamante, Peter Cohn, Cholsoon Jang, and Xia Yang. 2021. "Disparate Metabolomic Responses to Fructose Consumption between Different Mouse Strains and the Role of Gut Microbiota" Metabolites 11, no. 6: 342. https://doi.org/10.3390/metabo11060342
APA StyleAhn, I.-S., Yoon, J., Diamante, G., Cohn, P., Jang, C., & Yang, X. (2021). Disparate Metabolomic Responses to Fructose Consumption between Different Mouse Strains and the Role of Gut Microbiota. Metabolites, 11(6), 342. https://doi.org/10.3390/metabo11060342