
Untargeted Lipidomic Profiling of Dry Blood Spots Using SFC-HRMS

 , and
, and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Method Development
2.1.1. Optimization of Lipid Class Separation by Supercritical Fluid Chromatography
2.1.2. Optimization of Lipid Class Detection
2.1.3. In-House Database Development
2.1.4. Linearity and Sensitivity
2.2. MS-DIAL Processing Workflow
2.3. Analysis of NIST Reference Plasma
2.4. Assessment of the New SFC Method Using the Dry Blood Spot (DBS) Method
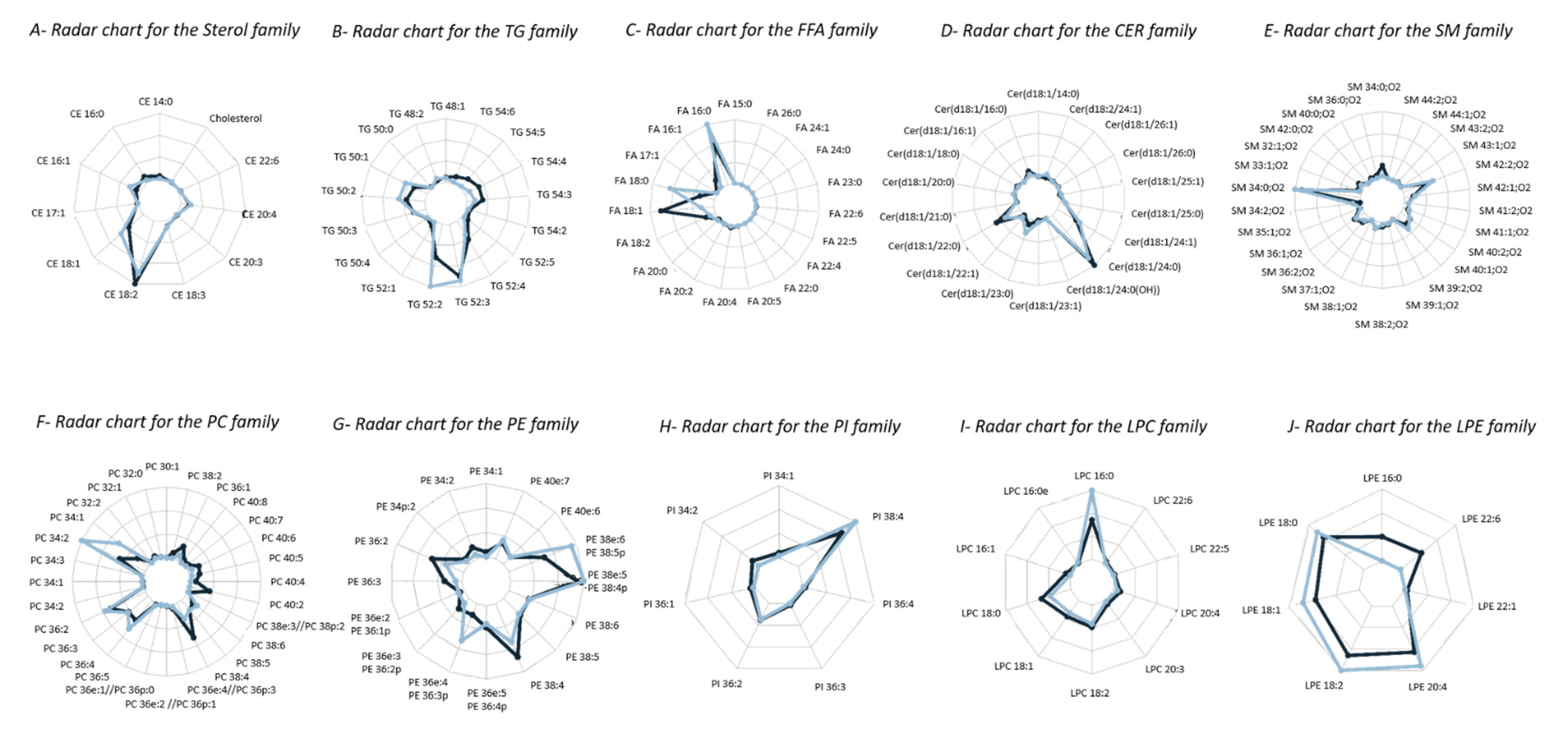
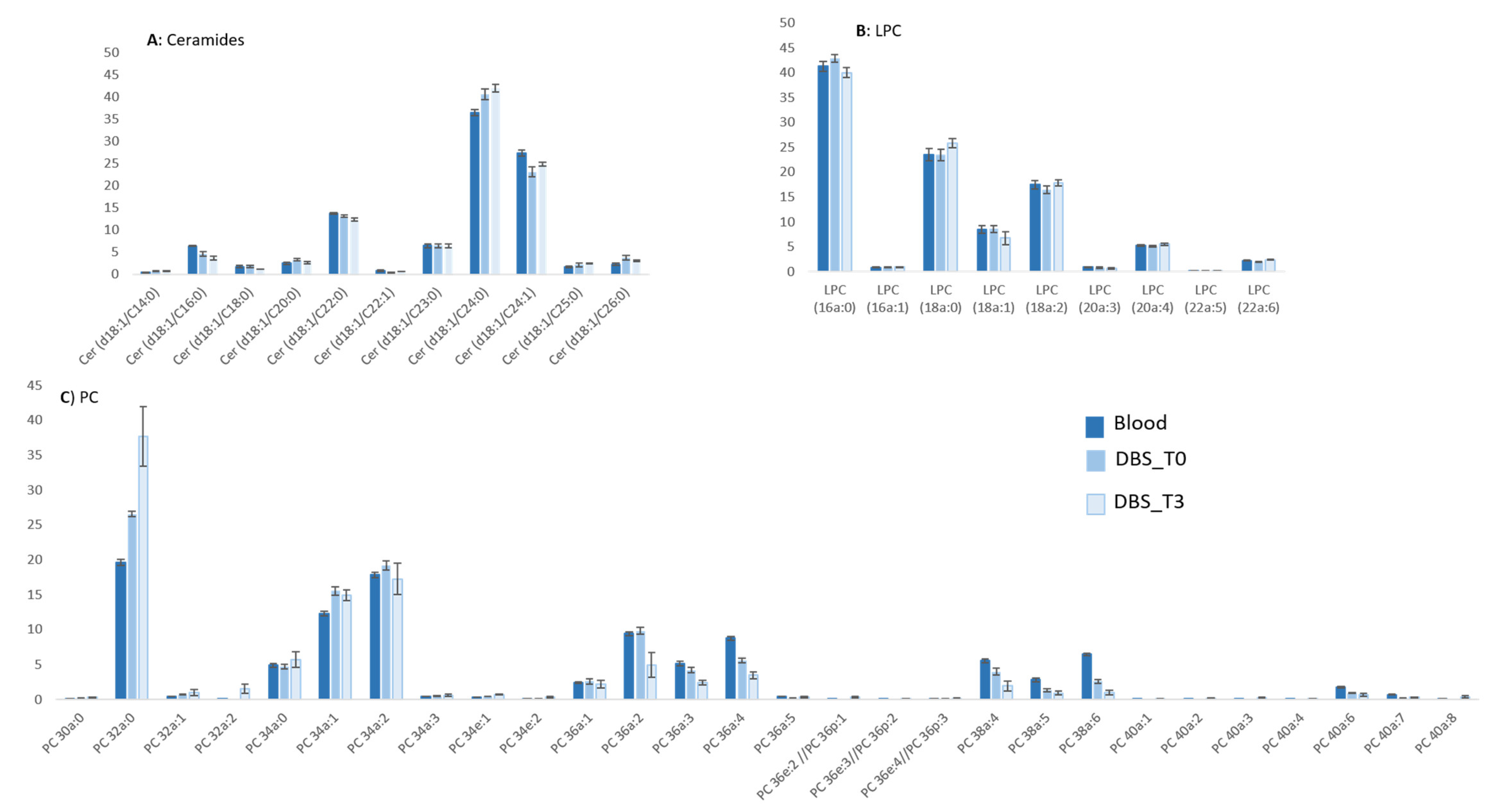
2.4.1. Lipidomic Profiling of Whole Blood vs. Dry Blood Spots
2.4.2. Storage Effect on Lipidomic Profiles
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Animals/DBS Prelevement
3.3. Sample Preparation
3.4. Supercritical Fluid Chromatography Conditions
3.5. Mass Spectrometry Parameters
3.6. Measurement of Linearity and Sensitivity
3.7. Data Analysis/Normalization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Hyötyläinen, T.; Orešič, M. Systems biology strategies to study lipidomes in health and disease. Prog. Lipid Res. 2014, 55, 43–60. [Google Scholar] [CrossRef]
- Wei, F.; Lamichhane, S.; Orešič, M.; Hyötyläinen, T. Lipidomes in health and disease: Analytical strategies and considerations. TrAC Trends Anal. Chem. 2019, 120, 115664. [Google Scholar] [CrossRef]
- Teo, C.C.; Chong, W.P.K.; Tan, E.; Basri, N.B.; Low, Z.J.; Ho, Y.S. Advances in sample preparation and analytical techniques for lipidomics study of clinical samples. TrAC Trends Anal. Chem. 2015, 66, 1–18. [Google Scholar] [CrossRef]
- Alves, M.A.; Lamichhane, S.; Dickens, A.; McGlinchey, A.; Ribeiro, H.C.; Sen, P.; Wei, F.; Hyötyläinen, T.; Orešič, M. Systems biology approaches to study lipidomes in health and disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2021, 1866, 158857. [Google Scholar] [CrossRef]
- Hu, C.; Duan, Q.; Han, X. Strategies to Improve/Eliminate the Limitations in Shotgun Lipidomics. Proteom. 2020, 20, e1900070. [Google Scholar] [CrossRef] [PubMed]
- Rampler, E.; Schoeny, H.; Mitic, B.M.; El Abiead, Y.; Schwaiger, M.; Koellensperger, G. Simultaneous non-polar and polar lipid analysis by on-line combination of HILIC, RP and high resolution MS. Anal. 2018, 143, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Lísa, M.; Holčapek, M. UHPSFC/ESI-MS Analysis of Lipids. Clinical 2018, 1730, 73–82. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, C.; Si, D.; Jia, B.; Zhong, L.; Yin, Y. Workflow development for targeted lipidomic quantification using parallel reaction monitoring on a quadrupole-time of flight mass spectrometry. Anal. Chim. Acta 2017, 972, 62–72. [Google Scholar] [CrossRef]
- Seyer, A.; Boudah, S.; Broudin, S.; Junot, C.; Colsch, B. Annotation of the human cerebrospinal fluid lipidome using high resolution mass spectrometry and a dedicated data processing workflow. Metabolomics 2016, 12, 91. [Google Scholar] [CrossRef]
- Yang, Y.; Liang, Y.; Yang, J.; Ye, F.; Zhou, T.; Gongke, L. Advances of supercritical fluid chromatography in lipid profiling. J. Pharm. Anal. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Chollet, C.; Boutet-Mercey, S.; Laboureur, L.; Rincon, C.; Méjean, M.; Jouhet, J.; Fenaille, F.; Colsch, B.; Touboul, D. Supercritical fluid chromatography coupled to mass spectrometry for lipidomics. J. Mass Spectrom. 2019, 54, 791–801. [Google Scholar] [CrossRef]
- Takeda, H.; Izumi, Y.; Takahashi, M.; Paxton, T.; Tamura, S.; Koike, T.; Yu, Y.; Kato, N.; Nagase, K.; Shiomi, M.; et al. Widely-targeted quantitative lipidomics method by supercritical fluid chromatography triple quadrupole mass spectrometry. J. Lipid Res. 2018, 59, 1283–1293. [Google Scholar] [CrossRef]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950–Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef]
- Kuipers, R.S.; Luxwolda, M.F.; Sango, W.S.; Kwesigabo, G.; Velzing-Aarts, F.V.; Dijck-Brouwer, D.J.; Muskiet, F.A. Postpartum changes in maternal and infant erythrocyte fatty acids are likely to be driven by restoring insulin sensitivity and DHA status. Med Hypotheses 2011, 76, 794–801. [Google Scholar] [CrossRef]
- Sabel, K.-G.; Lundqvist-Persson, C.; Bona, E.; Petzold, M.; Strandvik, B. Fatty acid patterns early after premature birth, simultaneously analysed in mothers’ food, breast milk and serum phospholipids of mothers and infants. Lipids Heal. Dis. 2009, 8, 20. [Google Scholar] [CrossRef]
- Lehmann, S.; Delaby, C.; Vialaret, J.; Ducos, J.; Hirtz, C. Current and Future Use of “Dried Blood Spot” Analyses in Clinical Chemistry. Clin. Chem. Lab. Med. 2013. [Google Scholar] [CrossRef]
- Lakshmy, R.; Tarik, M.; Abraham, R.A. Role of dried blood spots in health and disease diagnosis in older adults. Bioanalysis 2014, 6, 3121–3131. [Google Scholar] [CrossRef] [PubMed]
- Bamba, T.; Shimonishi, N.; Matsubara, A.; Hirata, K.; Nakazawa, Y.; Kobayashi, A.; Fukusaki, E. High throughput and exhaustive analysis of diverse lipids by using supercritical fluid chromatography-mass spectrometry for metabolomics. J. Biosci. Bioeng. 2008, 105, 460–469. [Google Scholar] [CrossRef]
- Lísa, M.; Holčapek, M. High-Throughput and Comprehensive Lipidomic Analysis Using Ultrahigh-Performance Supercritical Fluid Chromatography–Mass Spectrometry. Anal. Chem. 2015, 87, 7187–7195. [Google Scholar] [CrossRef]
- Murphy, R.C.; Axelsen, P.H. Mass spectrometric analysis of long-chain lipids. Mass Spectrom. Rev. 2010, 30, 579–599. [Google Scholar] [CrossRef] [PubMed]
- Sullards, M.C.; Liu, Y.; Chen, Y.; Merrill, A.H. Analysis of mammalian sphingolipids by liquid chromatography tandem mass spectrometry (LC-MS/MS) and tissue imaging mass spectrometry (TIMS). Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2011, 1811, 838–853. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. MS-DIAL 4: Accelerating Lipidomics Using an MS/MS, CCS, and Retention Time Atlas. bioRxiv 2020. [Google Scholar] [CrossRef]
- Khoury, S.; Canlet, C.; Lacroix, M.Z.; Berdeaux, O.; Jouhet, J.; Bertrand-Michel, J. Quantification of Lipids: Model, Reality, and Compromise. Biomolecules 2018, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Koulman, A.; Prentice, P.; Wong, M.C.Y.; Matthews, L.; Bond, N.J.; Eiden, M.; Griffin, J.L.; Dunger, D.B. The development and validation of a fast and robust dried blood spot based lipid profiling method to study infant metabolism. Metabolomics 2014, 10, 1018–1025. [Google Scholar] [CrossRef]
- Demirev, P.A. Dried Blood Spots: Analysis and Applications. Anal. Chem. 2013, 85, 779–789. [Google Scholar] [CrossRef]
- McDade, T.W.; Williams, S.; Snodgrass, J.J. What a drop can do: Dried blood spots as a minimally invasive method for integrating biomarkers into population-based research. Demography 2007, 44, 899–925. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- Folch, J.; Ascoli, I.; Lees, M.; A Meath, J.; LeBaron, N. Preparation of lipide extracts from brain tissue. J. Biol. Chem. 1951, 191, 833–841. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid Class | Species | RT (min) | Adduct | m/z Exp | m/z Th | Mass Dev (ppm) | m/z Daug Ion Exp | m/z Daug Ion Th | Mass Dev (ppm) | Structure of Daug Ion |
|---|---|---|---|---|---|---|---|---|---|---|
| Choesteryl ester | CE 17:0 | 0.94 | [M+Na]+ | 661.5886 | 661.5894 | 1.2 | no frag | |||
| Free cholesterol | Chol | 2.4 | [(M+H)-H2O]+ | 369.3522 | 369.3516 | −1.6 | 147.1152 | 147.1138 | −9.5 | C11H15+ (ring A and B) |
| Triglycerides | TG(17:0-17:1-17:0) (d5) | 1.05 | [M+NH4]+ | 869.8320 | 869.8329 | 1.0 | 582.5522 | 582.5496 | −4.5 | [(M+H)-FA]+ |
| TG 46:0 | 1.04 | [M+NH4]+ | 796.7406 | 796.7389 | −2.2 | 523.4724 | 523.4721 | −0.6 | [(M+H)-FA]+ | |
| Diglycerides | DG(12:0/12:0) | 2.19 | [(M+H)-H2O]+ | 439.3789 | 439.3782 | −1.6 | 183.1751 | 183.1748 | −1.6 | [(FA+H)-H2O]+ |
| DG 32:0 | 2.33 | [(M+H)-H2O]+ | 551.5043 | 551.5034 | −1.6 | 239.2376 | 239.2375 | −0.4 | [(FA+H)-H2O]+ | |
| Free fatty acid | FA 17:0 | 3.01 | [M-H]− | 269.2494 | 269.2486 | −3.0 | no frag | |||
| Ceramides | Cer(d18:1/12:0) | 3.19 | [(M+H)-H2O]+ | 464.4469 | 464.4462 | −1.5 | 264.2642 | 264.2682 | 15.1 | [(Sphingosine+H)-2H2O]+ |
| Cer(d18:1/24:0) | 3.27 | [(M+H)-H2O]+ | 632.6313 | 632.6340 | 4.3 | 264.2642 | 264.2682 | 15.1 | [(Sphingosine+H)-2H2O]+ | |
| Phosphatidylcholine | PC(13:0/13:0) | 4.36 | [M+H]+ | 650.4772 | 650.4755 | −2.6 | 184.072 | 184.0726 | 3.3 | phosphocholine ion |
| PC(16:0/18:1) | 4.34 | [M+H]+ | 760.5847 | 760.5851 | 0.5 | 184.072 | 184.0726 | 3.3 | phosphocholine ion | |
| Mono Hexosyl Ceramide | GlcCer(d18:1/12:0) | 4.58 | [(M+H)-H2O]+ | 626.4973 | 626.4990 | 2.7 | 264.2673 | 264.2682 | 3.4 | [(Sphingosine+H)-2H2O]+ |
| GalCer(d18:1/16:0) | 4.52 | [M+H]+ | 700.5751 | 700.5722 | −4.1 | 264.2707 | 264.2682 | −9.5 | [(Sphingosine+H)-2H2O]+ | |
| Sphingomyelin | SM(d18:1/12:0) | 4.68 | [M+H]+ | 647.5108 | 647.5123 | 2.2 | 184.072 | 184.0726 | 3.3 | phosphocholine ion |
| SM(d18:1/18:0) | 4.66 | [M+H]+ | 731.6033 | 731.6062 | 3.9 | 184.072 | 184.0726 | 3.3 | phosphocholine ion | |
| Fatty AcylCarnitine | CAR 12:0 | 4.78 | [M+H]+ | 344.2791 | 344.2795 | 1.2 | 183.1752 | 183.1748 | −2.2 | [(FA+H)-H2O]+ |
| Lysophosphatidylcholine | LPC 11:0 | 4.93 | [M+H]+ | 426.2642 | 426.2615 | −6.3 | 184.072 | 184.0726 | 3.3 | phosphocholine ion |
| LPC 20:0 | 4.83 | [M+H]+ | 552.4035 | 552.4024 | −2.1 | 184.072 | 184.0726 | 3.3 | phosphocholine ion | |
| Phosphatidylethanolamine | PE(12:0/12:0) | 5.21 | [M+H]+ | 580.3982 | 580.3973 | −1.6 | 439.3797 | 439.3782 | −3.4 | [(M+H)-phosphoethanolamine -H2O]+ |
| PE(16:0/16:0) | 5.29 | [M+H]+ | 692.5219 | 692.5225 | 0.9 | 551.5028 | 551.5034 | 1.1 | [(M+H)-phosphoethanolamine -H2O]+ | |
| Di Hexosyl Ceramide | LacCer(d18:1/12:0) | 5.54 | [M+H]+ | 806.5635 | 806.5624 | −1.3 | 264.2707 | 264.2682 | −9.5 | [(Sphingosine+H)-2H2O]+ |
| Lysophosphatidylethanolamine | LPE 13:0 | 6.21 | [M+H]+ | 412.2485 | 412.2459 | −6.4 | 271.2288 | 271.2268 | −7.4 | [(M-H)-ethanolamine]− |
| Phosphatidylglycerol | PG(12:0/12:0) | 6.6 | [M-H]− | 609.3776 | 609.3773 | −0.5 | 199.1721 | 199.1704 | −8.5 | RCOO− |
| Phosphatidylinositol | PI(15:0/18:1) (d7) | 7.84 | [M-H]− | 828.5634 | 828.5625 | −1.1 | 288.2911 | 288.2919 | 2.8 | RCOO− |
| PI(18:1/18:1) | 7.89 | [M-H]− | 861.5508 | 861.5499 | −1.0 | 281.2503 | 281.2486 | −6.0 | RCOO− | |
| Phosphatidylserine | PS(12:0/12:0) | 8.25 | [M+H]+ | 622.3727 | 622.3726 | −0.2 | 535.3327 | 535.3405 | 14.6 | [(M-H)-serine]− |
| PS(18:0/18:0) | 7.92 | [M-H]− | 790.5517 | 790.5604 | 11.0 | 703.5201 | 703.5283 | 11.7 | [(M-H)-serine]− |
| Metabolites | Repeatability (RSD, %) | Retention Time Variation (RSD, %) | Linearity r² | linearity (pg/µL) |
|---|---|---|---|---|
| Cholesterol | 2.6 | 0.4 | 0.997 | 3906.25–250,000 |
| CE 17:0 | 41.1 | 1.3 | 0.94 | 250–1953 |
| DG(12:0/12:0) | 4.2 | 0.7 | 0.98 | 250–3906 |
| TG(17:0/17:1/17:0d5) | 6.1 | 1.3 | 0.99 | 250–7813 |
| Cer(d18:1/12:0) | 13.4 | 0.2 | 0.99 | 250–7813 |
| LPC 11:0 | 11.0 | 0.2 | 0.97 | 250–31,250 |
| PC(13:0/13:0) | 5.4 | 0.0 | 0.99 | 250–31,250 |
| SM(d18:1/12:0) | 8.2 | 0.2 | 0.99 | 250–31,250 |
| GlcCer(d18:1/12:0) | 6.7 | 0.1 | 0.99 | 250–31,250 |
| CAR 12:0 | 27.0 | 0.0 | 0.966 | 250–31,250 |
| FA 17:0 | 23.6 | 0.0 | 0.99 | 250–31,250 |
| LacCer(d18:1/12:0) | 2.2 | 0.3 | 0.99 | 250–62,500 |
| PE(12:0/12:0) | 6.0 | 0.2 | 0.99 | 250–125,000 |
| LPE 13:0 | 0.8 | 0.2 | 0.99 | 250–125,000 |
| PG(12:0/12:0) | 10.0 | 0.1 | 0.99 | 250–125,000 |
| PI(15:0/18:1d7) | 2.1 | 0.1 | 0.99 | 250–125,000 |
| PS(12:0/12:0) | 4.6 | 0.2 | 0.99 | 25–125,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Faouder, P.; Soullier, J.; Tremblay-Franco, M.; Tournadre, A.; Martin, J.-F.; Guitton, Y.; Carlé, C.; Caspar-Bauguil, S.; Denechaud, P.-D.; Bertrand-Michel, J. Untargeted Lipidomic Profiling of Dry Blood Spots Using SFC-HRMS. Metabolites 2021, 11, 305. https://doi.org/10.3390/metabo11050305
Le Faouder P, Soullier J, Tremblay-Franco M, Tournadre A, Martin J-F, Guitton Y, Carlé C, Caspar-Bauguil S, Denechaud P-D, Bertrand-Michel J. Untargeted Lipidomic Profiling of Dry Blood Spots Using SFC-HRMS. Metabolites. 2021; 11(5):305. https://doi.org/10.3390/metabo11050305
Chicago/Turabian StyleLe Faouder, Pauline, Julia Soullier, Marie Tremblay-Franco, Anthony Tournadre, Jean-François Martin, Yann Guitton, Caroline Carlé, Sylvie Caspar-Bauguil, Pierre-Damien Denechaud, and Justine Bertrand-Michel. 2021. "Untargeted Lipidomic Profiling of Dry Blood Spots Using SFC-HRMS" Metabolites 11, no. 5: 305. https://doi.org/10.3390/metabo11050305
APA StyleLe Faouder, P., Soullier, J., Tremblay-Franco, M., Tournadre, A., Martin, J.-F., Guitton, Y., Carlé, C., Caspar-Bauguil, S., Denechaud, P.-D., & Bertrand-Michel, J. (2021). Untargeted Lipidomic Profiling of Dry Blood Spots Using SFC-HRMS. Metabolites, 11(5), 305. https://doi.org/10.3390/metabo11050305