
25CN-NBOMe Metabolites in Rat Urine, Human Liver Microsomes and C. elegans—Structure Determination and Synthesis of the Most Abundant Metabolites


 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Untargeted Screening and Detection Of Metabolites
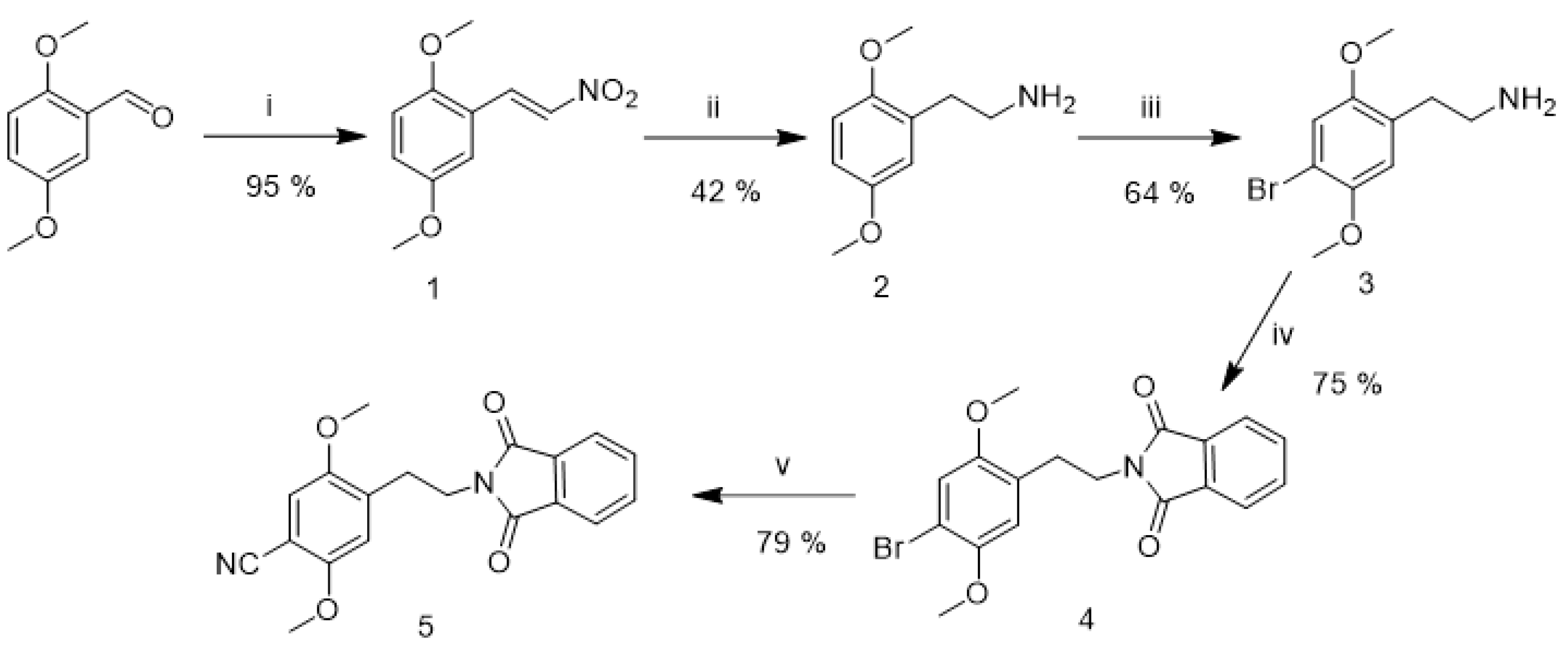
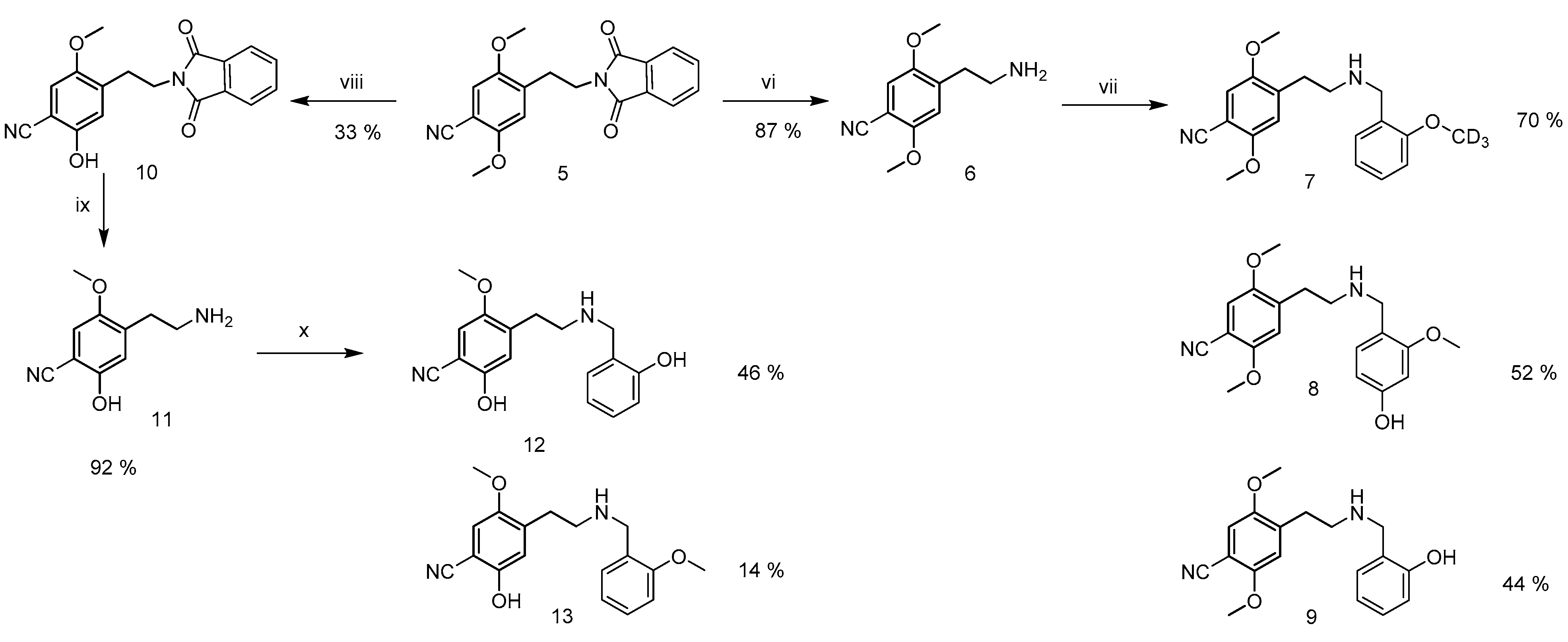
2.2. Synthesis of Reference Standards
2.3. LC-MS Analysis of the Most Abundant Metabolites
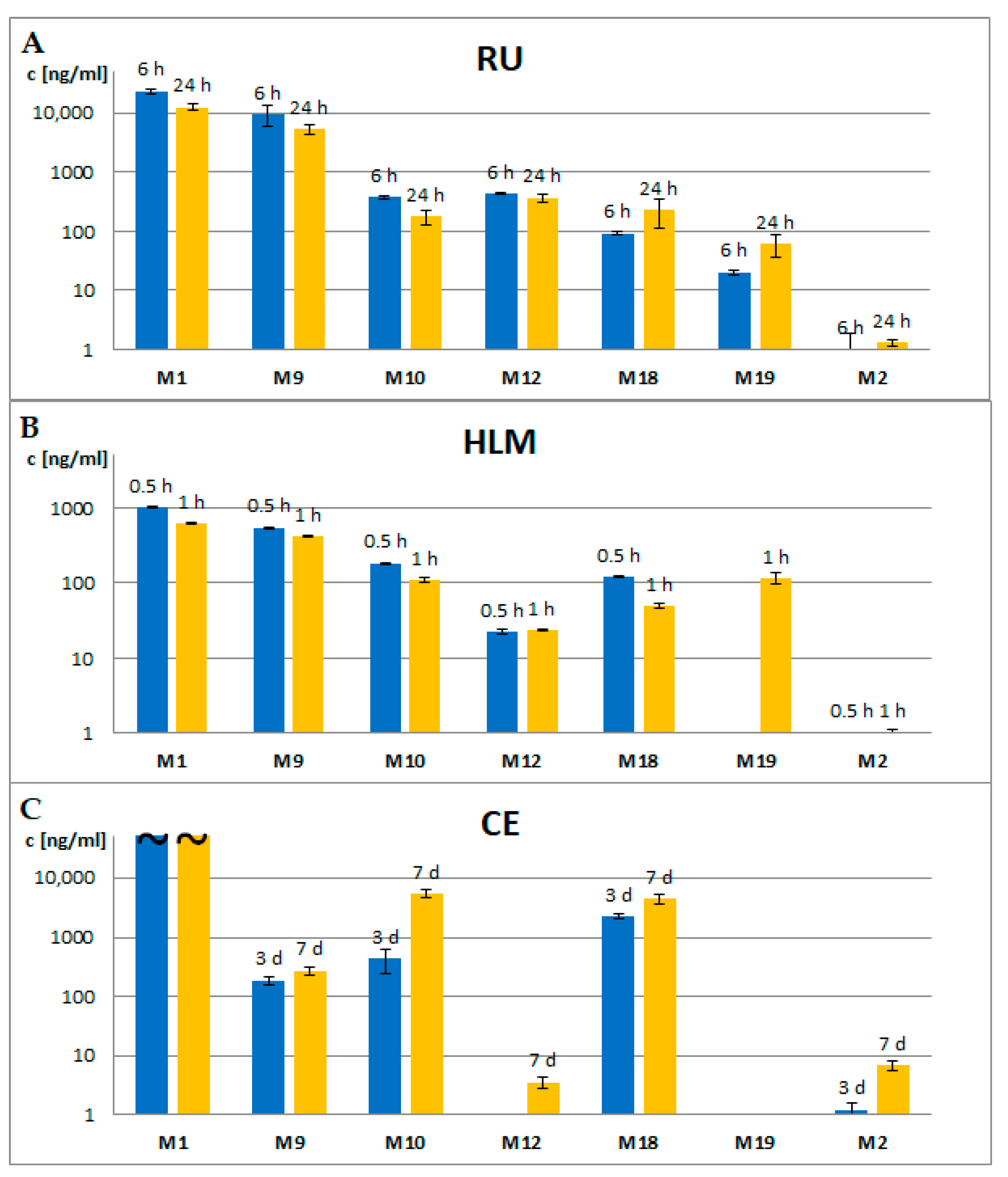
2.4. Comparison of Metabolite Formation In Vitro and In Vivo and in Different Species
2.5. Proposed Metabolic Pathways
3. Materials and Methods
3.1. In Vitro Incubations with Human Liver Microsomes
3.2. In Vitro Metabolism by Cunninghamella Elegans
3.3. Animals
3.4. Synthesis and Characterization of the Most Abundant Metabolites
3.4.1. General Information
3.4.2. Experimental Details
3.5. Sample Preparation
3.5.1. Human Liver Microsomes
3.5.2. C. elegans Mycelium & Culture Medium
3.5.3. Rat Urine
3.6. LC-MS Analysis
3.6.1. Untargeted Screening
3.6.2. Human Liver Microsomes and C. elegans Samples
3.6.3. Rat urine Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glennon, R.A.; Dukat, M.; El-Bermawy, M.; Law, H.; Angeles, J.D.L.; Teitler, M.; King, A.; Herrick-Davis, K. Influence of amine substituents on 5-HT2A versus 5-HT2C binding of phenylalkyl- and indolylalkylamines. J. Med. Chem. 1994, 37, 1929–1935. [Google Scholar] [CrossRef]
- Ettrup, A.; Hansen, M.; Santini, M.A.; Paine, J.; Gillings, N.; Palner, M.; Lehel, S.; Herth, M.M.; Madsen, J.; Kristensen, J.; et al. Radiosynthesis and in vivo evaluation of a series of substituted 11 C-phenethylamines as 5-HT 2A agonist PET tracers. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 681–693. [Google Scholar] [CrossRef]
- Ettrup, A.; Da Cunha-Bang, S.; McMahon, B.; Lehel, S.; Dyssegaard, A.; Skibsted, A.W.; Jørgensen, L.M.; Hansen, M.; O Baandrup, A.; Bache, S.; et al. Serotonin 2A Receptor Agonist Binding in the Human Brain with [11C]Cimbi-36. Br. J. Pharmacol. 2014, 34, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M. Design and Synthesis of Selective Serotonin Receptor Agonists for Positron Emission Tomography Imaging of the Brain. Ph.D. Thesis, University of Copenhagen, Copenhagen, Switzerland, 2010. [Google Scholar]
- Hansen, M.; Phonekeo, K.; Paine, J.S.; Leth-Petersen, S.; Begtrup, M. Synthesis and structure−activity relationships of N‑Benzyl phenethylamines as 5-HT2A/2C agonists. ACS Chem. Neurosci. 2014, 5, 243–249. [Google Scholar] [CrossRef]
- Jensen, A.A.; McCorvy, J.D.; Leth-Petersen, S.; Bundgaard, C.; Liebscher, G.; Kenakin, T.P.; Bräuner-Osborne, H.; Kehler, J.; Kristensen, J.L. Detailed characterization of the in vitro pharmacological and pharmacokinetic properties of N-(2-hydroxybenzyl)-2, 5-dimethoxy-4-cyanophenylethylamine (25CN-NBOH), a highly selective and brain-penetrant 5-HT2A receptor agonist. J. Pharmacol. Exp. Ther. 2017, 361, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Zuba, D.; Sekuła, K.; Buczek, A. 25C-NBOMe—New potent hallucinogenic substance identified on the drug market. Forensic Sci. Int. 2013, 227, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Poklis, J.L.; Raso, S.A.; Alford, K.N.; Poklis, A.; Peace, M.R. Analysis of 25I-NBOMe, 25B-NBOMe, 25C-NBOMe and other dimethoxyphenyl-N-[(2-Methoxyphenyl)Methyl] ethanamine derivatives on blotter paper. J. Anal. Toxicol. 2015, 39, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L. Pharmacology and toxicology of N-benzylphenethylamine (“NBOMe”) hallucinogens. Curr. Top. Behav. Neurosci. 2017, 32, 283–311. [Google Scholar] [CrossRef] [PubMed]
- Glennon, R.A.; Titeler, M.; McKenney, J.D. Evidence for 5-HT2 involvement in the mechanism of action of hallucinogenic agents. Life Sci. 1984, 35, 2505–2511. [Google Scholar] [CrossRef]
- Titeler, M.; Lyon, R.A.; Glennon, R.A. Radioligand binding evidence implicates the brain 5-HT2 receptor as a site of action for LSD and phenylisopropylamine hallucinogens. Psychopharmacology 1988, 94, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.X.; Vollenweider-Scherpenhuyzen, M.F.I.; Bäbler, A.; Vogel, H.; Hell, D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport 1998, 9, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Quednow, B.B.; Kometer, M.; Geyer, M.A.; Vollenweider, F.X. Psilocybin-induced deficits in automatic and controlled inhibition are attenuated by ketanserin in healthy human volunteers. Neuropsychopharmacology 2012, 37, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Valle, M.; Maqueda, A.E.; Rabella, M.; Rodríguez-Pujadas, A.; Antonijoan, R.M.; Romero, S.; Alonso, J.F.; Mañanas, M.À.; Barker, S.; Friedlander, P.; et al. Inhibition of alpha oscillations through serotonin-2A receptor activation underlies the visual effects of ayahuasca in humans. Eur. Neuropsychopharmacol. 2016, 26, 1161–1175. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L. Recent advances in the neuropsychopharmacology of serotonergic hallucinogens. Behav. Brain Res. 2014, 277, 99–120. [Google Scholar] [CrossRef] [PubMed]
- Pertz, H.H.; Heim, R.; Elz, S. N-Benzylated phenylethanamines are highly potent partial agonists at 5-HT2A receptors (abstract). Arch. Pharm. Pharm. Med. Chem. 2000, 333, 30. [Google Scholar]
- Braden, M.R.; Parrish, J.C.; Naylor, J.C.; Nichols, D.E. Molecular interaction of serotonin 5-HT 2A receptor residues phenethylamine agonists. Mol. Pharmacol. 2006, 70, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Eshleman, A.J.; Wolfrum, K.M.; Reed, J.F.; Kim, S.O.; Johnson, R.A.; Janowsky, A. Neurochemical pharmacology of psychoactive substituted N-benzylphenethylamines: High potency agonists at 5-HT2A receptors. Biochem Pharmacol. 2018, 158, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Heim, R. Synthese und Pharmakologie Potenter 5-HT2A-Rezeptoragonisten mit N-2-Methoxybenzyl-Partialstruktur. Ph.D. Thesis, Fachbereich Biologie, Chemie Pharmazie der Freien Universität, Berlin, Germany, 25 March 2003. [Google Scholar]
- Elmore, J.S.; Decker, A.M.; Sulima, A.; Rice, K.C.; Partilla, J.S.; Blough, B.E.; Baumann, M.H.; Carolina, N.; Section, S. Comparative neuropharmacology of N-(2-methoxybenzyl)-2,5-dimethoxyphenethylamine (NBOMe) hallucinogens and their 2C counterparts in male rats. Neuropharmacology 2019, 142, 240–250. [Google Scholar] [CrossRef]
- Rickli, A.; Luethi, D.; Reinisch, J.; Buchy, D.; Hoener, M.C.; Liechti, M.E. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology 2015, 99, 546–553. [Google Scholar] [CrossRef]
- Halberstadt, A.L.; Geyer, M.A. Characterization of the head-twitch response induced by hallucinogens in mice. Detection of the behavior based on the dynamics of head movement. Psychopharmacology 2013, 227, 727–739. [Google Scholar] [CrossRef]
- Canal, C.E.; Morgan, D. Head-twitch response in rodents induced by the hallucinogen 2,5-dimethoxy-4-iodoamphetamine: A comprehensive history, a re-evaluation of mechanisms, and its utility as a model. Drug Test. Anal. 2012, 4, 556–576. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Chatha, M.; Klein, A.K.; Wallach, J.; Brandt, S.D. Correlation between the potency of hallucinogens in the mouse head-twitch response assay and their behavioral and subjective effects in other species. Neuropharmacology 2020, 167, 107933. [Google Scholar] [CrossRef] [PubMed]
- Ettrup, A.; Holm, S.; Hansen, M.; Wasim, M.; Santini, M.A.; Palner, M.; Madsen, J.; Svarer, C.; Kristensen, J.L.; Knudsen, G.M. Preclinical safety assessment of the 5-HT2A receptor agonist PET radioligand [11C]Cimbi-36. Mol. Imaging Biol. 2013, 15, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Geyer, M.A. Effects of the hallucinogen 2,5-dimethoxy-4-iodophenethylamine (2C-I) and superpotent N-benzyl derivatives on the head twitch response. Neuropharmacology 2014, 77, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Poklis, J.L.; Poklis, A. “My friend said it was good LSD”: A suicide attempt following analytically confirmed 25I-NBOMe ingestion. J. Psychoact. Drugs 2014, 46, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Poklis, J.L.; Devers, K.G.; Arbefeville, E.F.; Pearson, J.M.; Houston, E.; Poklis, A. Postmortem detection of 25I-NBOMe [2-(4-iodo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine] in fluids and tissues determined by high performance liquid chromatography with tandem mass spectrometry from a traumatic death. Forensic Sci. Int. 2013, 234, e14–e20. [Google Scholar] [CrossRef] [PubMed]
- Poklis, J.L.; Dempsey, S.K.; Liu, K.; Ritter, J.K.; Wolf, C.; Zhang, S.; Poklis, A. Identification of metabolite biomarkers of the designer hallucinogen 25I-NBOMe in mouse hepatic microsomal preparations and human urine samples associated with clinical intoxication. J. Anal. Toxicol. 2015, 39, 607–616. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wohlfarth, A.; Roman, M.; Andersson, M.; Kugelberg, F.C.; Diao, X.; Carlier, J.; Eriksson, C.; Wu, X.; Konradsson, P.; Josefsson, M.; et al. 25C-NBOMe and 25I-NBOMe metabolite studies in human hepatocytes, in vivo mouse and human urine with high-resolution mass spectrometry. Drug Test. Anal. 2017, 9, 680–698. [Google Scholar] [CrossRef]
- Caspar, A.T.; Helfer, A.G.; Michely, J.A.; Auwärter, V.; Brandt, S.D.; Meyer, M.R.; Maurer, H.H. Studies on the metabolism and toxicological detection of the new in psychoactive designer drug 2-(4-iodo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine (25I-NBOMe) human and rat urine using GC-MS, LC-MS n, and LC-HR-MS/MS. Anal. Bioanal. Chem. 2015, 407, 6697–6719. [Google Scholar] [CrossRef] [PubMed]
- Caspar, A.T.; Brandt, S.D.; Stoever, A.E.; Meyer, M.R.; Maurer, H.H. Metabolic fate and detectability of the new psychoactive substances 2-(4-bromo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine (25B-NBOMe) and 2-(4-chloro-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine (25C-NBOMe) in human and rat urine by GC-MS, LC-MS n, and LC-HR-MS/MS approaches. J. Pharm. Biomed. Anal. 2017, 134, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.M.; Holm, B.; Leth-Petersen, S.; Kristensen, J.L.; Linnet, K. Characterization of the hepatic cytochrome P450 enzymes involved in the metabolism of 25I-NBOMe and 25I-NBOH. Drug Test Anal. 2016, 9, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Boumrah, Y.; Humbert, L.; Phanithavong, M. In vitro characterization of potential CYP- and UGT-derived metabolites of the psychoactive drug 25B-NBOMe using LC-high resolution MS. Drug Test Anal. 2016, 8, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Leth-Petersen, S.; Gabel-jensen, C.; Gillings, N.; Lehel, S.; Hansen, H.D.; Knudsen, G.M.; Kristensen, J.L. Metabolic fate of hallucinogenic NBOMes. Chem. Res. Toxicol. 2016, 26, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Kim, I.S.; Kim, Y.; Yoo, H.H.; Hong, J. Metabolic profile determination of 25N-NBOMe in human liver microsomes by liquid chromatography-quadrupole time-of-flight mass spectrometry. Int. J. Legal Med. 2019, 133, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Caspar, A.T.; Meyer, M.R.; Westphal, F.; Weber, A.A.; Maurer, H.H. Nano liquid chromatography-high-resolution mass spectrometry for the identification of metabolites of the two new psychoactive substances N-(ortho-methoxybenzyl)-3,4-dimethoxyamphetamine and N-(ortho-methoxybenzyl)-4-methylmethamphetamine. Talanta 2018, 188, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Grafinger, K.E.; Stahl, K.; Wilke, A.; König, S.; Weinmann, W. In vitro phase I metabolism of three phenethylamines 25D-NBOMe, 25E-NBOMe and 25N-NBOMe using microsomal and microbial models. Drug Test Anal. 2018, 10, 1607–1626. [Google Scholar] [CrossRef]
- Ketha, H.; Webb, M.; Clayton, L.; Li, S. Gas Chromatography Mass Spectrometry (GC-MS) for Identification of Designer Stimulants Including 2C Amines, NBOMe Compounds, and Cathinones in Urine. Curr. Protoc. Toxicol. 2017, 74, 4.43.1–4.43.10. [Google Scholar] [CrossRef] [PubMed]
- Richter, L.H.J.; Maurer, H.H.; Meyer, M.R. New psychoactive substances. Studies on the metabolism of XLR-11, AB-PINACA, FUB-PB-22, 4-methoxy-α-PVP, 25-I-NBOMe, and meclonazepam using human liver preparations in comparison to primary human hepatocytes, and human urine. Toxicol. Lett. 2017, 280, 142–150. [Google Scholar] [CrossRef]
- Temporal, K.H.; Scott, K.S.; Mohr, A.L.A.; Logan, B.K. Metabolic profile determination of NBOMe compounds using human liver microsomes and comparison with findings in authentic human blood and urine. J. Anal. Toxicol. 2017, 41, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Caspar, A.A.T.; Meyer, M.R.; Maurer, H.H. Human cytochrome P450 kinetic studies on six N-2-methoxybenzyl (NBOMe)-derived new psychoactive substances using the substrate depletion approach. Toxicol. Lett. 2017, 285, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leth-Petersen, S.; Bundgaard, C.; Hansen, M.; Carnerup, M.A.; Kehler, J.; Kristensen, J.L. Correlating the metabolic stability of psychedelic 5-HT 2A agonists with anecdotal reports of human oral bioavailability. Neurochem. Res. 2014, 39, 2018–2023. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.L.; Collings, B.A.; Le Blanc, J.C.Y.; Hager, J.W. A novel MS3 experiment for quantifying ions with a linear ion trap. Can. J. Chem. 2018, 96, 653–663. [Google Scholar] [CrossRef]
- Markus, B.; Kwon, C. In vitro metabolism of aromatic nitriles. J. Pharm. Sci. 1994, 83, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- ThermoFisher. Available online: https://www.thermofisher.com/cz/en/home/references/protocols/drug-discovery/adme-tox-protocols/microsomes-protocol.html (accessed on 17 March 2021).
- Chemistry Archive. Available online: https://erowid.org/archive/rhodium/chemistry/edda.html (accessed on 4 January 2021).
- Butterick, J.R.; Unrau, A.M. Reduction of β-nitrostyrene with sodium bis-(2-methoxyethoxy)-aluminium dihydride. A convenient route to substituted phenylisopropylamines. J. Chem. Soc. Chem. Commun. 1974, 8, 307–308. [Google Scholar] [CrossRef]
- Xu, Y.-Z.; Chen, C. Synthesis of deuterium labeled phenethylamine derivatives. J. Label Compd. Radiopharm. 2006, 49, 1187–2000. [Google Scholar] [CrossRef]
- Cheng, A.C.; Castagnoli, N. Synthesis and physicochemical and neurotoxicity studies of l-(4-substituted-2,5-dihydroxyphenyl)-2-aminoethane analogues of 6-hydroxydopamine. J. Med. Chem. 1984, 27, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Hájková, K.; Jurásek, B.; Čejka, J.; Štefková, K.; Páleníček, T.; Sýkora, D.; Kuchař, M. Synthesis and identification of deschloroketamine metabolites in rats’ urine and a quantification method for deschloroketamine and metabolites in rats’ serum and brain tissue using liquid chromatography tandem mass spectrometry. Drug Test. Anal. 2020, 12, 343–360. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No. | Metabolite | Precursor ion Exact Mass [m/z] | RT [min] | RU | HLM | CE |
|---|---|---|---|---|---|---|
| M1 | 25CN-NBOMe | 327.1709 | 7.9 | I | I | I |
| M2 | hydroxy-25CN-NBOMe isomer 1 | 343.1658 | 7.4 | I | ||
| M3 | hydroxy-25CN-NBOMe isomer 2 | 343.1658 | 7.3 | I | I | I |
| M4 | hydroxy-25CN-NBOMe isomer 3 | 343.1658 | 7.7 | D | ||
| M5 | dehydro-25CN-NBOMe | 325.1552 | 7.3 | D | I | |
| M6 | dehydro-25CN-NBOMe-N-oxide | 341.1501 | 9.8 | I | D | |
| M7 | dehydro-hydroxy-25CN-NBOMe | 341.1501 | 10.1 | D | I | |
| M8 | O-demethyl-hydroxy-25CN-NBOMe | 329.1501 | 6.9 | D | I | |
| M9 | O-demethyl-25CN-NBOMe | 313.1552 | 7.5 | I | I | I |
| M10 | 25CN-NBOH | 313.1552 | 7.6 | I | I | I |
| M11 | O,O-bis-demethyl-25CN-NBOMe | 299.1396 | 6.8 | I | ||
| M12 | O-demethyl-25CN-NBOH | 299.1396 | 7.1 | I | I | D |
| M13 | O,O-bis-demethyl-hydroxy-25CN-NBOMe isomer 1 | 315.1345 | 5.8 | D | ||
| M14 | O,O-bis-demethyl-hydroxy-25CN-NBOMe isomers 2, 3, 4 | 315.1345 | 6.1–6.8 | D | ||
| M15 | O-demethyl-dehydro-hydroxy-25CN-NBOMe | 327.1345 | 9.2 | D | ||
| M16 | O,O-bis-demethyl-dehydro-25CN-NBOMe | 297.1239 | 6.9 | D | ||
| M17 | O,O-bis-demethyl-25CN-NBOH | 285.1239 | 5.7 | D | ||
| M18 | 2C-CN | 207.1134 | 5.7 | I | I | I |
| M19 | O-demethyl-2C-CN | 193.0977 | 4.9 | I | D | |
| M20 | O-demethyl-deamino-hydroxy-2C-CN | 194.0817 | 6.0 | D | ||
| M21 | O-demethyl-deamino-carboxy-2C-CN | 208.0610 | 5.7 | I | ||
| M22 | 2C-CONH2-NBOMe | 345.1814 | 7.1 | D | ||
| M23 | O-demethyl-2C-CONH2 | 211.1078 | 4.0. | D | ||
| M24 | O,O-bis-demethyl-hydroxy-2C-COOH-NBOMe | 334.1291 | 5.4 | D | ||
| M25 | 2C-COOH | 226.1079 | 4.5 | D | ||
| M8I G | O-demethyl-hydroxy-25CN-NBOMe glucuronide isomer 1 | 505.1822 | 6.2 | I | ||
| M8 G | O-demethyl-hydroxy-25CN-NBOMe glucuronide isomer 2 | 505.1822 | 6.7 | D | ||
| M9 G | O-demethyl-25CN-NBOMe glucuronide | 489.1873 | 6.9 | I | D | |
| M11 G | O,O-bis-demethyl-25CN-NBOMe glucuronide | 475.1717 | 6.1 | I | D | |
| M13 G | O,O-bis-demethyl-hydroxy-25CN-NBOMe glucuronide isomer 1 | 491.1666 | 5.4 | D | ||
| M14 G | O,O-bis-demethyl-hydroxy-25CN-NBOMe glucuronide isomer 2 | 491.1666 | 6.2 | D | ||
| M16 G | O,O-bis-demethyl-dehydro-25CN-NBOMe glucuronide | 473.1560 | 5.6 | D | ||
| M17 G | O,O-bis-demethyl-25CN-NBOH glucuronide | 461.1560 | 5.4 | I | ||
| M19 G | O-demethyl-2C-CN glucuronide | 369.1298 | 4.0 | D | ||
| M26 G | oxo-2C-CN glucuronide | 397.1247 | 4.5 | D | ||
| M3 S | hydroxy-25CN-NBOMe sulfate | 423.1226 | 7.4 | D | ||
| M8 S | O-demethyl-hydroxy-25CN-NBOMe sulfate | 409.1069 | 6.9 | I | ||
| M9 S | O-demethyl-25CN-NBOMe sulfate | 393.112 | 7.7 | D | ||
| M11 S | O,O-bis-demethyl-25CN-NBOMe sulfate | 379.0964 | 6.9 | I | ||
| M12 S | O-demethyl-25CN-NBOH sulfate | 379.0964 | 7.1 | D | ||
| M13 S | O,O-bis-demethyl-hydroxy-25CN-NBOMe sulfate isomer 1 | 395.0913 | 5.8 | D | ||
| M14 S1 | O,O-bis-demethyl-hydroxy-25CN-NBOMe sulfate isomer 2 | 395.0913 | 6.3 | D | ||
| M14 S2 | O,O-bis-demethyl-hydroxy-25CN-NBOMe sulfate isomer 3 | 395.0913 | 6.8 | D | ||
| M17 S1 | O,O-bis-demethyl-25CN-NBOH sulfate isomer 1 | 365.0807 | 5.7 | D | ||
| M17 S2 | O,O-bis-demethyl-25CN-NBOH sulfate isomer 2 | 365.0807 | 6.4 | D | ||
| M26 S | oxo-2C-CN sulfate | 301.0494 | 5.0 | I | ||
| M19 Ac | O-demethyl-2C-CN N-acetyl isomer 1 | 235.1083 | 7.1 | I | ||
| M19 Ac | O-demethyl-2C-CN N-acetyl isomer 2 | 235.1083 | 7.3 | I | ||
| M19 Ac G | O-demethyl-2C-CN N-acetyl glucuronide | 411.1404 | 5.9 | I | ||
| M19 Ac S | O-demethyl-2C-CN N-acetyl sulfate | 315.0651 | 6.2 | D | ||
| M27 Ac | hydroxy-2C-CN N-acetyl | 265.1188 | 5.6 | D |
| No. | Target Metabolite | Precursor Ion [m/z] | MS2 Fragment Ions [m/z] 1 | MS3 Fragment Ions [m/z] 1 | RT [min] | Detected in Sample |
|---|---|---|---|---|---|---|
| M1 | 25CN-NBOMe | 327 | 77 (1), 91 (37), 93 (13), 121 (100), 205 (2) | 121: 77 (8), 91 (100), 93 (83), 121 (60) 205: 175 (31), 190 (79), 205 (100) | 8.3 | RU, HLM, CE |
| M2 | hydroxy-25CN-NBOMe 1 (8) | 343 | 107 (8), 137 (100), 175 (1), 190 (30), 207 (30) | 190: 163 (1), 175 (21), 190 (100) 207: 147 (1), 160 (3), 163 (1), 175 (100) | 7.8 | RU, HLM, CE |
| M9 | O-demethyl-25CN-NBOMe (13) | 313 | 91 (14), 93 (6), 121 (100), 150 (7), 312 (9) | 91: 65 (100), 91 (65), 93 (97) 121: 77 (8), 91 (88), 93 (100), 121 (34) | 7.9 | RU, HLM, CE |
| M10 | 25CN-NBOH (9) | 313 | 77 (44), 79 (50), 106 (100), 160 (5), 175 (78), 190 (98), 207 (85), 313 (7) | 190: 160 (5), 175 (100), 190 (3) 207: 190 (100), 175 (10), 207 (2) | 8.0 | RU, HLM, CE |
| M12 | O-demethyl-25CN-NBOH (12) | 299 | 77 (4), 79 (5), 107 (42), 161 (2), 176 (67), 193 (100), 299 (22) | 176: 121 (1), 148 (1), 149 (2), 161 (16), 176 (100) 193: 149 (2), 161 (14), 176 (100), 193 (93) | 7.5 | RU, HLM, CE |
| M18 | 2C-CN (6) | 207 | 105 (5), 133 (6), 147 (7), 160 (23), 175 (100), 190 (85) | 175: 104 (1), 132 (1), 160 (100), 175 (16) 190: 117 (1), 147 (1), 160 (5), 175 (100), 190 (2) | 6.3 | RU, HLM, CE |
| M19 | O-demethyl-2C-CN (11) | 193 | 91 (2), 118 (3), 121 (2), 133 (4), 148 (10), 161 (92), 176 (100) | 161: 132 (2), 160 (14), 161 (100) 176: 148 (2), 149 (2), 161 (15), 176 (100) | 5.5 | RU, HLM |
| 1st Precursor [m/z] | 2nd Precursor [m/z] | Quantifier/Qualifier Ion [m/z] | CE 1 [eV] | DP 2 [V] | |
|---|---|---|---|---|---|
| 25CN-NBOMe | 327.2 | 121.1 | 93.0 | 19 | 9 |
| 327.2 | 205.1 | 190.1 | 15 | 42 | |
| O-demethyl-25CN-NBOMe (13) | 313.2 | 121.1 | 93.0 | 21 | 30 |
| 313.2 | 91.0 | 65.0 | 64 | 21 | |
| 25CN-NBOH (9) | 313.2 | 207.2 | 190.1 | 17 | 90 |
| 313.2 | 190.1 | 175.1 | 21 | 17 | |
| O-demethyl-25CN-NBOH (12) | 299.2 | 176.1 | 161.1 | 22 | 65 |
| 299.2 | 193.1 | 176.1 | 17 | 60 | |
| 2C-CN (6) | 207.2 | 175.1 | 160.1 | 26 | 50 |
| 207.2 | 190.1 | 175.1 | 14 | 30 | |
| O-demethyl-2C-CN (11) | 193.1 | 176.1 | 161.1 | 15 | 15 |
| 193.1 | 161.1 | 132.1 | 28 | 39 | |
| hydroxy-25CN-NBOMe (8) | 343.3 | 207.2 | 190.1 | 12 | 36 |
| 343.3 | 190.1 | 175.1 | 17 | 21 | |
| 25CN-NBOMe-d3 (7) | 330.2 | 124.1 | 96.0 | 24 | 22 |
| 330.2 | 205.1 | 190.1 | 15 | 42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šuláková, A.; Nykodemová, J.; Palivec, P.; Jurok, R.; Rimpelová, S.; Leonhardt, T.; Šíchová, K.; Páleníček, T.; Kuchař, M. 25CN-NBOMe Metabolites in Rat Urine, Human Liver Microsomes and C. elegans—Structure Determination and Synthesis of the Most Abundant Metabolites. Metabolites 2021, 11, 212. https://doi.org/10.3390/metabo11040212
Šuláková A, Nykodemová J, Palivec P, Jurok R, Rimpelová S, Leonhardt T, Šíchová K, Páleníček T, Kuchař M. 25CN-NBOMe Metabolites in Rat Urine, Human Liver Microsomes and C. elegans—Structure Determination and Synthesis of the Most Abundant Metabolites. Metabolites. 2021; 11(4):212. https://doi.org/10.3390/metabo11040212
Chicago/Turabian StyleŠuláková, Anna, Jitka Nykodemová, Petr Palivec, Radek Jurok, Silvie Rimpelová, Tereza Leonhardt, Klára Šíchová, Tomáš Páleníček, and Martin Kuchař. 2021. "25CN-NBOMe Metabolites in Rat Urine, Human Liver Microsomes and C. elegans—Structure Determination and Synthesis of the Most Abundant Metabolites" Metabolites 11, no. 4: 212. https://doi.org/10.3390/metabo11040212
APA StyleŠuláková, A., Nykodemová, J., Palivec, P., Jurok, R., Rimpelová, S., Leonhardt, T., Šíchová, K., Páleníček, T., & Kuchař, M. (2021). 25CN-NBOMe Metabolites in Rat Urine, Human Liver Microsomes and C. elegans—Structure Determination and Synthesis of the Most Abundant Metabolites. Metabolites, 11(4), 212. https://doi.org/10.3390/metabo11040212