
Calibration-Curve-Locking Database for Semi-Quantitative Metabolomics by Gas Chromatography/Mass Spectrometry

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
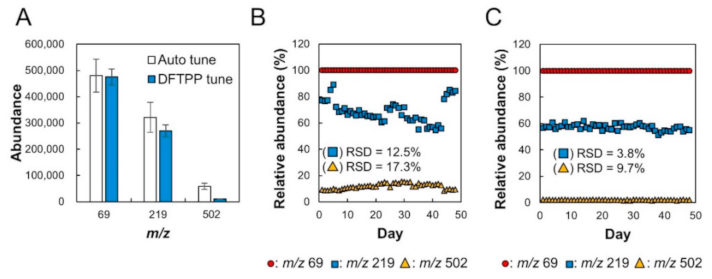
2.1. Verification of Stability of Relative Sensitivity of Mass Spectrometry
2.2. Optimization of Automatic Derivatization Condition Using PAL RTC
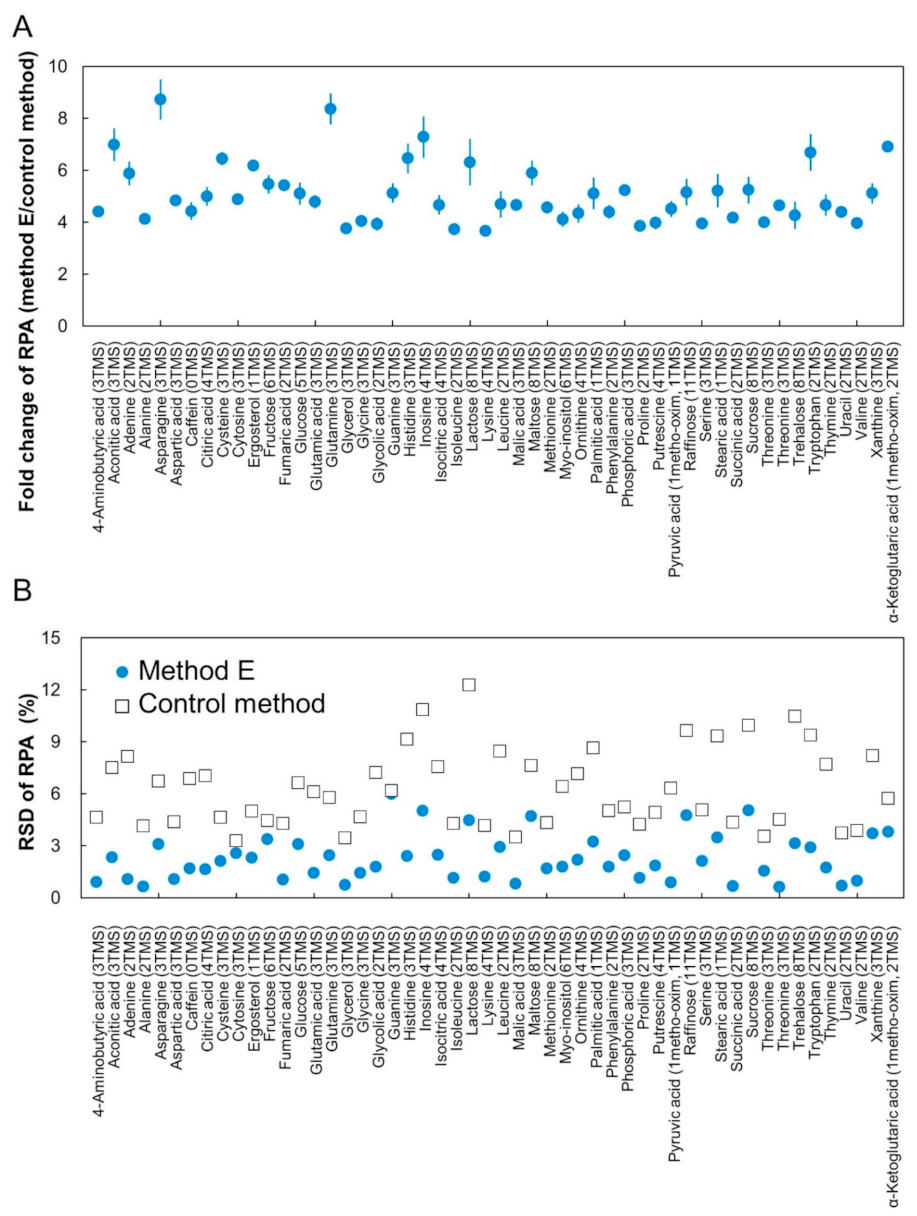
2.3. Construction of CCLD
2.4. Method Validation by Quantification of Human Plasma Sample
3. Materials and Methods
3.1. Material and Reagents
3.2. Preparation of Standard Mixtures
3.3. Extraction of Metabolites from the Plasma Sample
3.4. Automated Derivatization and GC/MS Analysis
3.5. Data Analysis
3.6. Preparation of Calibration Curves
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Büscher, J.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Siskos, A.P.; Jain, P.; Römisch-Margl, W.; Bennett, M.; Achaintre, D.; Asad, Y.; Marney, L.; Richardson, L.; Koulman, A.; Griffin, J.L.; et al. Interlaboratory Reproducibility of a Targeted Metabolomics Platform for Analysis of Human Serum and Plasma. Anal. Chem. 2017, 89, 656–665. [Google Scholar] [CrossRef]
- Izumi, Y.; Matsuda, F.; Hirayama, A.; Ikeda, K.; Kita, Y.; Horie, K.; Saigusa, D.; Saito, K.; Sawada, Y.; Nakanishi, H.; et al. Inter-Laboratory Comparison of Metabolite Measurements for Metabolomics Data Integration. Metabolites 2019, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 2009, 5, 435–458. [Google Scholar] [CrossRef]
- Noack, S.; Wiechert, W. Quantitative metabolomics: A phantom? Trends Biotechnol. 2014, 32, 238–244. [Google Scholar] [CrossRef]
- Papadimitropoulos, M.-E.P.; Vasilopoulou, C.G.; Maga-Nteve, C.; Klapa, M.I. Untargeted GC-MS Metabolomics; Humana Press: New York, NY, USA, 2018; pp. 133–147. [Google Scholar]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [PubMed]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmuller, E.; Dormann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. GMD@CSB.DB: The Golm Metabolome Database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef]
- Tsugawa, H.; Tsujimoto, Y.; Arita, M.; Bamba, T.; Fukusaki, E. GC/MS based metabolomics: Development of a data mining system for metabolite identification by using soft independent modeling of class analogy (SIMCA). BMC Bioinform. 2011, 12, 1–13. [Google Scholar] [CrossRef]
- Schauer, N.; Steinhauser, D.; Strelkov, S.; Schomburg, D.; Allison, G.; Moritz, T.; Lundgren, K.; Roessner-Tunali, U.; Forbes, M.G.; Willmitzer, L.; et al. GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 2005, 579, 1332–1337. [Google Scholar] [CrossRef]
- Ishida, T.; Kudo, K.; Naka, S.; Toubou, K.; Noguchi, T.; Ikeda, N. Rapid diagnosis of drug intoxication using novel NAGINATA TM gas chromatography/mass spectrometry software. Rapid Commun. Mass Spectrom. 2007, 21, 3129–3138. [Google Scholar] [CrossRef]
- Kudo, K.; Usumoto, Y.; Usui, K.; Hayashida, M.; Kurisaki, E.; Saka, K.; Tsuji, A.; Ikeda, N. Rapid and simultaneous extraction of acidic and basic drugs from human whole blood for reliable semi-quantitative NAGINATA drug screening by GC–MS. Forensic Toxicol. 2014, 32, 97–104. [Google Scholar] [CrossRef]
- Kadokami, K.; Tanada, K.; Taneda, K.; Nakagawa, K. Novel gas chromatography-mass spectrometry database for automatic identification and quantification of micropollutants. J. Chromatogr. A 2005, 1089, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kanani, H.H.; Klapa, M.I. Data correction strategy for metabolomics analysis using gas chromatography-mass spectrometry. Metab. Eng. 2007, 9, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kanani, H.; Chrysanthopoulos, P.K.; Klapa, M.I. Standardizing GC-MS metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Quéro, A.; Jousse, C.; Lequart-Pillon, M.; Gontier, E.; Guillot, X.; Courtois, B.; Courtois, J.; Pau-Roblot, C. Improved stability of TMS derivatives for the robust quantification of plant polar metabolites by gas chromatography-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 970, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; David, F.; Lynen, F.; Rumpel, K.; Dugardeyn, J.; Van Der Straeten, D.; Xu, G.; Sandra, P. Evaluation of automated sample preparation, retention time locked gas chromatography-mass spectrometry and data analysis methods for the metabolomic study of Arabidopsis species. J. Chromatogr. A 2011, 1218, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Khakimov, B.; Motawia, M.S.; Bak, S.; Engelsen, S.B. The use of trimethylsilyl cyanide derivatization for robust and broad-spectrum high-throughput gas chromatography-mass spectrometry based metabolomics. Anal. Bioanal. Chem. 2013, 405, 9193–9205. [Google Scholar] [CrossRef] [PubMed]
- Zarate, E.; Boyle, V.; Rupprecht, U.; Green, S.; Villas-Boas, S.G.; Baker, P.; Pinu, F.R. Fully automated trimethylsilyl (TMS) derivatisation protocol for metabolite profiling by GC-MS. Metabolites 2017, 7, 1. [Google Scholar] [CrossRef]
- Erban, A.; Schauer, N.; Fernie, A.R.; Kopka, J. Nonsupervised Construction and Application of Mass Spectral and Retention Time Index Libraries From Time-of-Flight Gas Chromatography-Mass Spectrometry Metabolite Profiles. Metabolomics 2006, 358, 19–39. [Google Scholar]
- Miyagawa, H.; Bamba, T. Comparison of sequential derivatization with concurrent methods for GC/MS-based metabolomics. J. Biosci. Bioeng. 2019, 127, 160–168. [Google Scholar] [CrossRef]
- Glick, N.; Fischer, M. Adipic aciduria, a dietary artefact. J. Inherit. Metab. Dis. 1991, 14, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, G.; Mutlu, H.; Ozturk, E.; Sildiroglu, H.O.; Keskin, A.T.; Basekim, C.C.; Kizilkaya, E. Magnetic resonance imaging findings of adult-onset glutaric aciduria type I. Acta Radiol. 2007, 48, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Liebich, H.M.; Pickert, A.; Stierle, U.; Wöll, J. Gas chromatography—Mass spectrometry of saturated and unsaturated dicarboxylic acids in urine. J. Chromatogr. A 1980, 199, 181–189. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | Metabolite | RT (min) | Quantification Ion | Qualifier Ion 1 | Qualifier Ion 2 | Slope | Intercept | R2 |
|---|---|---|---|---|---|---|---|---|
| Mean ± SD (n = 9) | (m/z) | (m/z) | (m/z) | (n = 9) | (n = 9) | |||
| M001 | 4-Aminobutyric acid (3TMS) | 13.31 ± 0.01 | 304 | 174 | 147 | 0.00428 | −0.01677 | 0.997946326 |
| M002 | Aconitic acid (3TMS) | 15.81 ± 0.01 | 147 | 375 | 229 | 0.00939 | −0.10858 | 0.971925122 |
| M003 | Adenine (2TMS) | 17.12 ± 0.01 | 264 | 279 | 192 | 0.0145 | −0.1298 | 0.985093709 |
| M004 | Alanine (2TMS) | 7.43 ± 0.01 | 116 | 190 | 147 | 0.0181 | −0.0537 | 0.998107977 |
| M005 | Asparagine (3TMS) | 14.93 ± 0.01 | 116 | 231 | 132 | 0.00296 | −0.03480 | 0.969363443 |
| M006 | Aspartic acid (3TMS) | 13.16 ± 0.01 | 232 | 218 | 100 | 0.0148 | −0.1094 | 0.990087236 |
| M007 | Caffeine (0TMS) | 17.01 ± 0.01 | 194 | 109 | 67 | 0.00655 | −0.04092 | 0.992869316 |
| M008 | Citric acid (4TMS) | 16.55 ± 0.01 | 273 | 465 | 347 | 0.0186 | −0.1197 | 0.993577885 |
| M009 | Cysteine (3TMS) | 13.6 ± 0.01 | 218 | 220 | 100 | 0.00555 | −0.04003 | 0.991182834 |
| M010 | Cytosine (3TMS) | 13.22 ± 0.01 | 254 | 240 | 170 | 0.00398 | −0.02034 | 0.995979412 |
| M011 | Ergosterol (1TMS) | 27.96 ± 0.01 | 211 | 364 | 129 | 0.00116 | −0.01162 | 0.979552269 |
| M012 | Fructose-syn (5TMS) | 17.07 ± 0.01 | 307 | 217 | 103 | 0.00309 | −0.00293 | 0.998573559 |
| M013 | Fumaric acid (2TMS) | 10.99 ± 0.01 | 245 | 147 | 73 | 0.0101 | −0.0890 | 0.984258298 |
| M014 | Glucose-syn (5TMS) | 17.33 ± 0.01 | 319 | 205 | 160 | 0.00777 | −0.04160 | 0.995802756 |
| M015 | Glutamic acid (3TMS) | 14.36 ± 0.01 | 246 | 147 | 128 | 0.00846 | −0.05905 | 0.991560137 |
| M016 | Glutamine (3TMS) | 16.09 ± 0.01 | 156 | 245 | 73 | 0.00282 | −0.03872 | 0.952720346 |
| M017 | Glycerol (3TMS) | 9.89 ± 0.01 | 205 | 147 | 117 | 0.00465 | −0.02928 | 0.989621885 |
| M018 | Glycine (3TMS) | 10.38 ± 0.01 | 174 | 248 | 73 | 0.0217 | −0.0472 | 0.999309039 |
| M019 | Glycolic acid (2TMS) | 7.04 ± 0.01 | 205 | 177 | 147 | 0.00119 | −0.01314 | 0.972607162 |
| M020 | Guanine (3TMS) | 19.59 ± 0.01 | 352 | 264 | 73 | 0.00705 | −0.06942 | 0.980404182 |
| M021 | Histidine (3TMS) | 17.61 ± 0.01 | 154 | 254 | 0 | 0.0133 | −0.1708 | 0.96381778 |
| M022 | Inosine (4TMS) | 23.31 ± 0.01 | 217 | 281 | 230 | 0.00624 | −0.08974 | 0.948511774 |
| M023 | Isocitric acid (4TMS) | 16.55 ± 0.01 | 245 | 319 | 204 | 0.00604 | −0.03097 | 0.996408844 |
| M024 | Isoleucine (2TMS) | 10.19 ± 0.01 | 158 | 232 | 218 | 0.0212 | −0.0629 | 0.998183005 |
| M025 | Lactose1 (8TMS) | 24.14 ± 0.01 | 204 | 361 | 319 | 0.0159 | −0.1570 | 0.98100185 |
| M026 | Leucine (2TMS) | 9.89 ± 0.01 | 158 | 232 | 73 | 0.0247 | −0.0639 | 0.99895856 |
| M027 | Lysine (4TMS) | 17.63 ± 0.01 | 174 | 317 | 230 | 0.0142 | −0.0658 | 0.997051812 |
| M028 | Malic acid (3TMS) | 12.75 ± 0.01 | 233 | 335 | 147 | 0.00342 | −0.02041 | 0.993892629 |
| M029 | Maltose2 (8TMS) | 24.7 ± 0.01 | 361 | 204 | 103 | 0.00214 | −0.02062 | 0.979669477 |
| M030 | Methionine (2TMS) | 13.17 ± 0.01 | 176 | 293 | 219 | 0.0129 | −0.0779 | 0.994154978 |
| M031 | Myo-inositol (6TMS) | 19.24 ± 0.01 | 305 | 265 | 191 | 0.0153 | −0.0500 | 0.998677671 |
| M032 | Ornithine (4TMS) | 16.55 ± 0.01 | 142 | 420 | 174 | 0.0219 | −0.0493 | 0.998580314 |
| M033 | Palmitic acid (1TMS) | 18.88 ± 0.01 | 117 | 313 | 129 | 0.00928 | −0.05299 | 0.992456268 |
| M034 | Phenylalanine (2TMS) | 14.47 ± 0.01 | 192 | 218 | 73 | 0.011 | −0.0526 | 0.996722448 |
| M035 | Phosphoric acid (3TMS) | 9.86 ± 0.01 | 299 | 314 | 211 | 0.013 | −0.1179 | 0.981330404 |
| M036 | Proline (2TMS) | 10.27 ± 0.01 | 142 | 216 | 73 | 0.0242 | −0.1219 | 0.995436881 |
| M037 | Putrescine (4TMS) | 15.72 ± 0.01 | 174 | 214 | 200 | 0.043 | −0.0501 | 0.999693324 |
| M038 | Pyruvic acid (1metho-oxim 1TMS) | 6.64 ± 0.01 | 174 | 115 | 89 | 0.000672 | −0.00155 | 0.999376913 |
| M039 | Raffinose (11TMS) | 28.81 ± 0.01 | 361 | 437 | 217 | 0.0228 | −0.1821 | 0.987840487 |
| M040 | Serine (3TMS) | 11.09 ± 0.01 | 204 | 278 | 73 | 0.0145 | −0.0565 | 0.99774593 |
| M041 | Stearic acid (1TMS) | 20.68 ± 0.01 | 117 | 341 | 145 | 0.00862 | −0.06043 | 0.98769425 |
| M042 | Succinic acid (2TMS) | 10.49 ± 0.01 | 147 | 129 | 73 | 0.0197 | −0.0733 | 0.996924908 |
| M043 | Sucrose (8TMS) | 23.75 ± 0.01 | 361 | 437 | 217 | 0.0218 | −0.1727 | 0.988630816 |
| M044 | Threonine (3TMS) | 11.43 ± 0.01 | 218 | 291 | 117 | 0.00732 | −0.02761 | 0.997716556 |
| M045 | Thymine (2TMS) | 11.64 ± 0.01 | 255 | 147 | 113 | 0.0113 | −0.0662 | 0.99383769 |
| M046 | Trehalose (8TMS) | 24.55 ± 0.01 | 191 | 217 | 103 | 0.0167 | −0.0683 | 0.997932624 |
| M047 | Tryptophan (2TMS) | 20.24 ± 0.01 | 218 | 130 | 100 | 0.0121 | −0.1471 | 0.970083386 |
| M048 | Tyrosine (3TMS) | 17.81 ± 0.01 | 218 | 280 | 179 | 0.0322 | −0.1704 | 0.996105297 |
| M049 | Uracil (2TMS) | 10.81 ± 0.01 | 241 | 99 | 147 | 0.00851 | −0.04013 | 0.99612372 |
| M050 | Valine (2TMS) | 9.09 ± 0.01 | 144 | 246 | 218 | 0.0202 | −0.0626 | 0.99818831 |
| M051 | Xanthine (3TMS) | 18.57 ± 0.01 | 353 | 368 | 147 | 0.004 | −0.03673 | 0.983502896 |
| M052 | α-Ketoglutaric acid | 13.84 ± 0.01 | 198 | 147 | 89 | 0.000811 | −0.007595 | 0.980432179 |
| (1_metho-oxim 2TMS) |
| ID | Metabolite | Recovery (%) | RSD (%) | ID | Metabolite | Recovery (%) | RSD (%) | ID | Metabolite | Recovery (%) | RSD (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M001 | 4-Aminobutyric acid (3TMS) | 93.8 | 5.6 | M019 | Glycolic acid (2TMS) | 120.2 | 5.7 | M037 | Putrescine (4TMS) | 89.0 | 6.9 |
| M002 | Aconitic acid (3TMS) | 67.1 | 5.0 | M020 | Guanine (3TMS) | 92.9 | 6.4 | M038 | Pyruvic acid (1metho-oxim 1TMS) | 114.9 | 21.1 |
| M003 | Adenine (2TMS) | 88.1 | 5.1 | M021 | Histidine (3TMS) | 102.3 | 5.7 | M039 | Raffinose (11TMS) | 133.1 | 8.2 |
| M004 | Alanine (2TMS) | 74.3 | 16.5 | M022 | Inosine (4TMS) | 98.4 | 9.0 | M040 | Serine (3TMS) | 88.6 | 8.7 |
| M005 | Asparagine (3TMS) | 61.1 | 6.4 | M023 | Isocitric acid (4TMS) | 90.9 | 11.0 | M041 | Stearic acid (1TMS) | 81.4 | 10.0 |
| M006 | Aspartic acid (3TMS) | 74.8 | 7.2 | M024 | Isoleucine (2TMS) | 79.1 | 7.4 | M042 | Succinic acid (2TMS) | 85.1 | 6.8 |
| M007 | Caffeine (0TMS) | 96.2 | 5.5 | M025 | Lactose1 (8TMS) | 109.7 | 9.7 | M043 | Sucrose (8TMS) | 123.8 | 9.2 |
| M008 | Citric acid (4TMS) | 92.7 | 12.0 | M026 | Leucine (2TMS) | 79.5 | 10.1 | M044 | Threonine (3TMS) | 92.0 | 8.5 |
| M009 | Cysteine (3TMS) | 82.8 | 3.9 | M027 | Lysine (4TMS) | 120.5 | 7.0 | M045 | Thymine (2TMS) | 76.3 | 10.7 |
| M010 | Cytosine (3TMS) | 92.5 | 7.6 | M028 | Malic acid (3TMS) | 82.3 | 5.6 | M046 | Trehalose (8TMS) | 123.0 | 10.1 |
| M011 | Ergosterol (1TMS) | 112.5 | 7.0 | M029 | Maltose2 (8TMS) | 125.5 | 11.0 | M047 | Tryptophan (2TMS) | 80.0 | 6.8 |
| M012 | Fructose-syn (5TMS) | 76.1 | 21.4 | M030 | Methionine (2TMS) | 86.0 | 4.2 | M048 | Tyrosine (3TMS) | 111.9 | 6.7 |
| M013 | Fumaric acid (2TMS) | 83.9 | 5.6 | M031 | Myo-inositol (6TMS) | 120.1 | 7.7 | M049 | Uracil (2TMS) | 79.4 | 8.2 |
| M014 | Glucose-syn (5TMS) | 104.0 | 10.3 | M032 | Ornithine (4TMS) | 99.5 | 7.7 | M050 | Valine (2TMS) | 68.6 | 15.1 |
| M015 | Glutamic acid (3TMS) | 106.7 | 5.9 | M033 | Palmitic acid (1TMS) | 83.0 | 9.7 | M051 | Xanthine (3TMS) | 58.3 | 5.9 |
| M016 | Glutamine (3TMS) | 129.9 | 10.0 | M034 | Phenylalanine (2TMS) | 96.7 | 5.7 | M052 | α-Ketoglutaric acid (1_metho-oxim 2TMS) | 99.1 | 5.4 |
| M017 | Glycerol (3TMS) | 110.3 | 14.2 | M035 | Phosphoric acid (3TMS) | 120.0 | 11.7 | ||||
| M018 | Glycine (3TMS) | 111.7 | 10.5 | M036 | Proline (2TMS) | 83.1 | 8.5 |
| ID | Metabolite | RT (min) | This Work | Literature a |
|---|---|---|---|---|
| Mean ± SD (n = 9) | (mmol/L in Plasma) (n = 9) | (mmol/L in Plasma) | ||
| M004 | Alanine (2TMS) | 7.42 ± 0.00 | 276 ± 21 | 300 ± 26 |
| M005 | Asparagine (3TMS) | 14.92 ± 0.00 | 71.3 ± 1.4 | - |
| M006 | Aspartic acid (3TMS) | 13.15 ± 0.00 | 39.5 ± 0.3 | - |
| M008 | Citric acid (4TMS) | 16.54 ± 0.00 | 48 ± 2.8 | - |
| M009 | Cysteine (3TMS) | 13.6 ± 0.00 | 41.8 ± 0.8 | 44.3 ± 6.9 |
| M012 | Fructose-syn (5TMS) | 17.06 ± 0.00 | 96.9 ± 20.1 | - |
| M014 | Glucose-syn (5TMS) | 17.33 ± 0.00 | 4270 ± 366 | 4560 ± 56 |
| M015 | Glutamic acid (3TMS) | 14.35 ± 0.00 | 71.5 ± 3.2 | 67.4 ± 18 |
| M016 | Glutamine (3TMS) | 16.08 ± 0.00 | 284 ± 11 | - |
| M017 | Glycerol (3TMS) | 9.89 ± 0.00 | 179 ± 13 | - |
| M018 | Glycine (3TMS) | 10.38 ± 0.00 | 143 ± 8 | 245 ± 16 |
| M021 | Histidine (3TMS) | 17.6 ± 0.00 | 93 ± 4.8 | 72.6 ± 3.6 |
| M024 | Isoleucine (2TMS) | 10.19 ± 0.00 | 59.5 ± 2 | 55.5 ± 3.4 |
| M026 | Leucine (2TMS) | 9.89 ± 0.00 | 105 ± 4 | 100 ± 6 |
| M027 | Lysine (4TMS) | 17.63 ± 0.00 | 80.1 ± 11.3 | 140 ± 14 |
| M030 | Methionine (2TMS) | 13.17 ± 0.00 | 40.7 ± 0.4 | 22.3 ± 1.8 |
| M031 | Myo-inositol (6TMS) | 19.23 ± 0.00 | 30.7 ± 1.5 | - |
| M032 | Ornithine (4TMS) | 16.55 ± 0.00 | 29 ± 2.7 | 52.1 ± 2.8 |
| M034 | Phenylalanine (2TMS) | 14.47 ± 0.00 | 53.4 ± 1.5 | 50.8 ± 7 |
| M035 | Phosphoric acid (3TMS) | 9.87 ± 0.00 | 275 ± 47 | - |
| M036 | Proline (2TMS) | 10.27 ± 0.00 | 158 ± 9 | 177 ± 9 |
| M038 | Pyruvic acid (1metho-oxim 1TMS) | 6.62 ± 0.00 | 283 ± 36 | - |
| M040 | Serine (3TMS) | 11.09 ± 0.00 | 73.1 ± 3.8 | 95.9 ± 4.3 |
| M042 | Succinic acid (2TMS) | 10.49 ± 0.00 | 25.6 ± 0.7 | - |
| M044 | Threonine (3TMS) | 11.43 ± 0.00 | 103 ± 7 | 119 ± 6 |
| M047 | Tryptophan (2TMS) | 20.23 ± 0.00 | 69 ± 2 | - |
| M048 | Tyrosine (3TMS) | 17.81 ± 0.00 | 57.5 ± 2.2 | 57.3 ± 3 |
| M050 | Valine (2TMS) | 9.09 ± 0.00 | 156 ± 10 | 182 ± 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hata, K.; Soma, Y.; Yamashita, T.; Takahashi, M.; Sugitate, K.; Serino, T.; Miyagawa, H.; Suzuki, K.; Yamada, K.; Kawamukai, T.; et al. Calibration-Curve-Locking Database for Semi-Quantitative Metabolomics by Gas Chromatography/Mass Spectrometry. Metabolites 2021, 11, 207. https://doi.org/10.3390/metabo11040207
Hata K, Soma Y, Yamashita T, Takahashi M, Sugitate K, Serino T, Miyagawa H, Suzuki K, Yamada K, Kawamukai T, et al. Calibration-Curve-Locking Database for Semi-Quantitative Metabolomics by Gas Chromatography/Mass Spectrometry. Metabolites. 2021; 11(4):207. https://doi.org/10.3390/metabo11040207
Chicago/Turabian StyleHata, Kosuke, Yuki Soma, Toshiyuki Yamashita, Masatomo Takahashi, Kuniyo Sugitate, Takeshi Serino, Hiromi Miyagawa, Kenichi Suzuki, Kayoko Yamada, Takatomo Kawamukai, and et al. 2021. "Calibration-Curve-Locking Database for Semi-Quantitative Metabolomics by Gas Chromatography/Mass Spectrometry" Metabolites 11, no. 4: 207. https://doi.org/10.3390/metabo11040207
APA StyleHata, K., Soma, Y., Yamashita, T., Takahashi, M., Sugitate, K., Serino, T., Miyagawa, H., Suzuki, K., Yamada, K., Kawamukai, T., Shiota, T., Izumi, Y., & Bamba, T. (2021). Calibration-Curve-Locking Database for Semi-Quantitative Metabolomics by Gas Chromatography/Mass Spectrometry. Metabolites, 11(4), 207. https://doi.org/10.3390/metabo11040207