1. Introduction
Metabolites are both downstream products and regulators of the majority of biological processes. The ability to quantify them in a nondestructive manner from a test sample offers us an opportunity to use them as a proxy to study or monitor these processes in a variety of biological systems, including live cell cultures. If automated, this would enable in vivo repeated inference of cell phenotype and its gene or protein interactions at regular time intervals, possibly in a high-throughput setup. Some high-throughput metabolomics applications include drug discovery [
1,
2,
3], toxicology [
4], biomass processing [
5], and vaccine production in bioreactors [
6,
7].
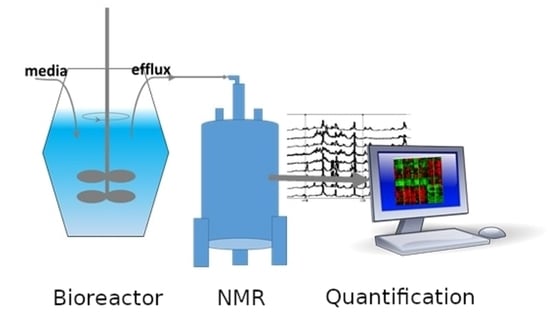
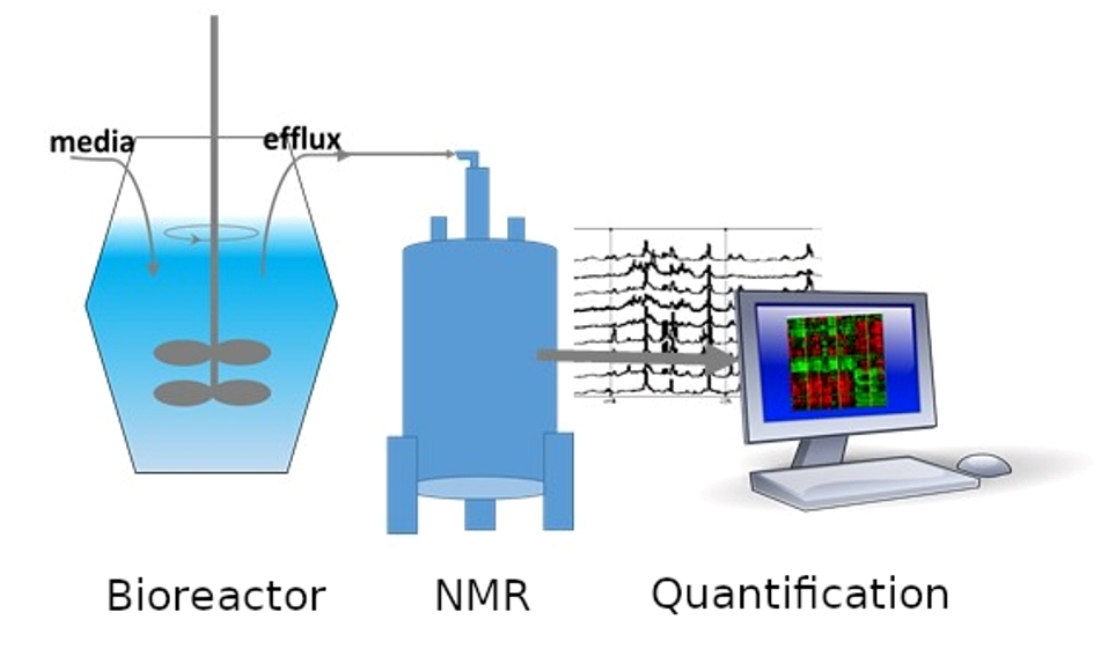
Nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) are presently the most popular methods to analyze metabolites. Although MS is more sensitive and can, with application of different MS approaches, detect more metabolites than NMR, NMR is attractive for quantitative metabolomics because NMR experiments can be performed easily and quickly without complicated sample preparation procedures. Following investment in an NMR instrument, experiments are inexpensive, and highly reproducible. MS experiments require more involved preparations, extensive quality control, and are destructive to the test sample. The nondestructive aspect of NMR makes it interesting as a first step in further, perhaps more time-consuming or expensive analysis as well as possible analysis of live cells. In bioreactor applications, NMR metabolomics is uniquely attractive for continual media monitoring, which can provide extensive data for condition optimization. Flow NMR probes allow continual sample measurement with no need for NMR sample tubes, or extensive sample preparation such as spectrometer shimming as only the sample solution itself is exchanged. Quantitative NMR metabolomics can be envisioned as an additional sensor for monitoring the molecules involved in the reactor process.
The most popular approach in NMR metabolomics, including quantitative NMR metabolomics, is the 1D
H NMR. Other nuclei, such as
C,
N, or
P can also be used in natural abundance or in isotopically labeled samples. The physics behind the 1D
H NMR relate the concentration of the metabolites’ signals in a straightforward manner [
8], and an experiment can be performed in a matter of minutes. We refer the reader to the review by Emwas et al. [
9] for more information on the different NMR methods and its comparison to MS for metabolomics.
The 1D NMR has a relatively narrow spectral range, and multidimensional NMR can better identify a large number of metabolites in a test sample. However, multidimensional NMR experiments generally take significantly longer to perform, and the concentration of each metabolite in the test sample could have different proportionality constants to the signal volume [
8,
9,
10]. Recent literature, such as [
10], provide strategies and protocols to mitigate the different proportionality constants and slow acquisition time issues, and quantification from 2D NMR seems attainable under certain conditions. Still, 1D NMR experiments remain a preferred method for most metabolomics applications due to its fast measurement time, which is a desirable characteristic in a monitoring application. There are also more 1D
H NMR metabolite standards in public databases than any other types of NMR, which is useful for the metabolite assignment and quantification task.
Presently, most metabolomic studies rely on manual or semimanual NMR spectra profiling, identification, or quantification by trained experts. This is because profiling many different metabolites from standard libraries remains an open pattern recognition problem. Although there are guides on this manual task (e.g., [
11]) and several automated and semiautomated methods have been published [
12,
13,
14,
15], a fully automated solution remains elusive. It would also be beneficial if such an algorithm could provide some form of uncertainty quantification to better inform downstream analysis algorithms. In addition, existing literature requires specific experimental conditions or experimental designs for accurate profiling or quantification of 1D
H NMR spectra; for example, a set of protocols were identified for plant metabolomics in [
16], blood profiles in [
17], celebrospinal fluid in [
18], urine in [
19], and experiment design methods were proposed for the bioreactor setting in [
20].
Most existing quantitative NMR literature focus on biofluids that are relevant for clinical applications of NMR metabolomics. Many such applications have test samples that arise from controlled environments, which are similar to the standards from public libraries [
9,
21,
22]. Conversely, bioreactors, generally defined as vessels where biological reaction or change is taking place, can involve enzymes (e.g., for cell-free bioprocessing), microorganisms (e.g., for environmental remediation or fermentation), animal cells (e.g., for production of biologics or vaccines), plant cells (e.g., for production of bioactive compounds), and tissues (e.g., for cell therapies). To provide a reliable yield in these types of bioreactor applications, it is crucial to maintain a suitable environment for the desired biological reaction to take place. This is only possible with sufficient information about the cellular environment at different time stamps of the reactor process.
Most existing quantifiers also focus on applications where the metabolites have mostly reached homeostasis, and that the metabolic profiles differ mostly in concentration but not composition, i.e., no new metabolites are being introduced between experiments. In the case where multiple experiments were conducted at different times during a particular run of a bioreactor, some of the experiment samples could contain different types of byproduct metabolites that are not present in the other samples. Therefore, methodologies for fast, high-throughput quantitative metabolomics of bioreactors require quantifiers that are robust to the challenges in this setting. We discuss these issues in
Section 3.
There are a few recent reviews that touch on quantitative NMR (e.g., [
9,
21,
22]). However, to the best of our knowledge, benchmark or performance analysis for existing quantitative NMR approaches applied to the bioreactor setting is lacking. We first review a popular 1D
H NMR signal model, followed by some challenges for quantifying metabolite concentrations. We then briefly review some existing NMR quantifiers before reporting our benchmark results. Our motivation for the benchmark section is to provide insight into how some existing NMR quantifiers [
12,
14,
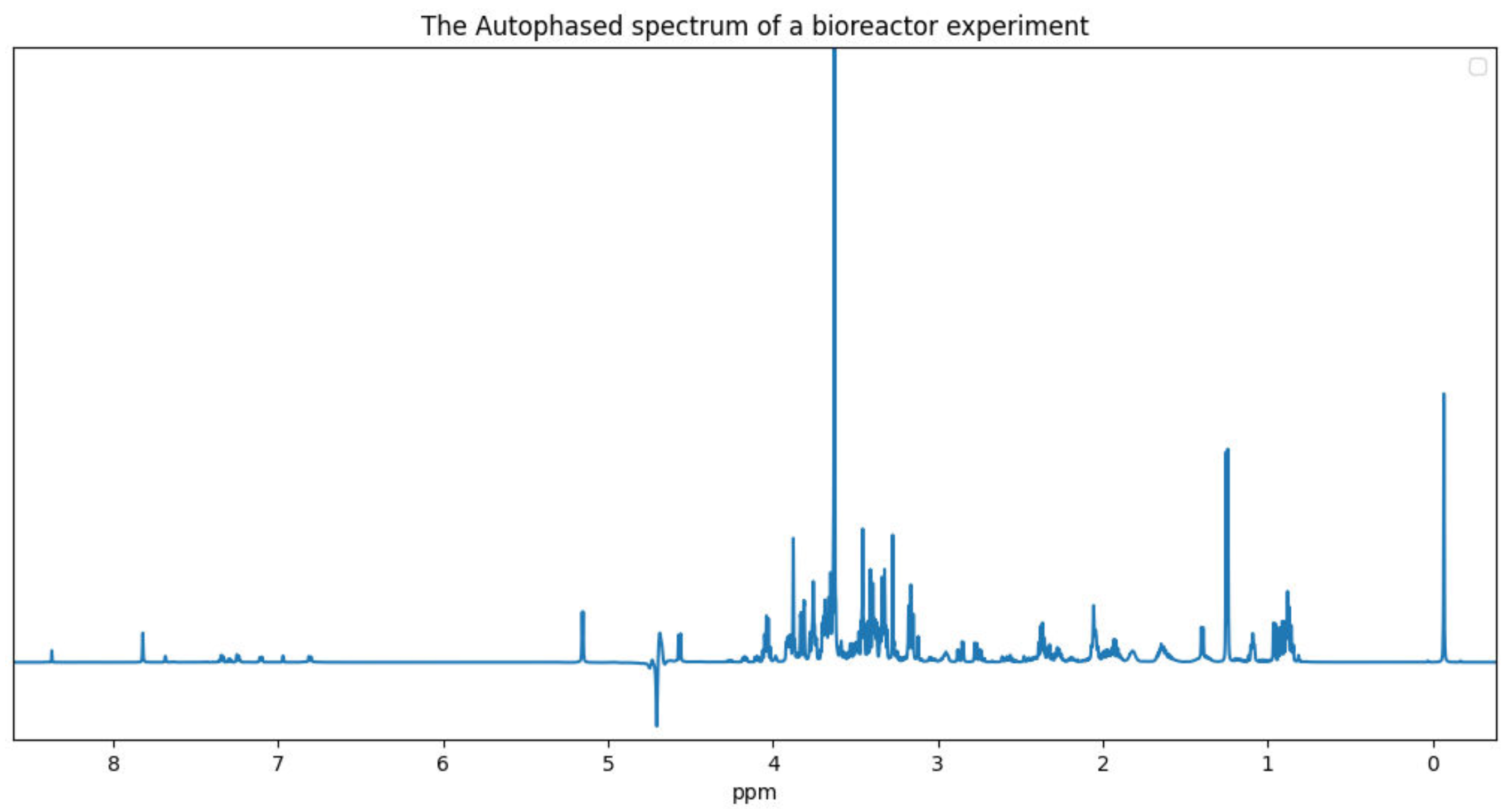
15] perform against our benchmark sample. These quantifiers were chosen because they are publicly accessible and are still being maintained. We used Dulbecco’s modified Eagle’s medium (DMEM), a cell growth medium, as our benchmark biological sample. The experiment associated with our benchmark data was conducted using a 600 megahertz (MHz) spectrometer. This cell medium was chosen because of the presence of glucose and a number of other amino acids that are commonly found in mammalian cell bioreactor applications. We conclude with some potential avenues of exploration for future quantifiers. The benchmark NMR data is included in the
supplementary material.
4. Existing Quantifiers
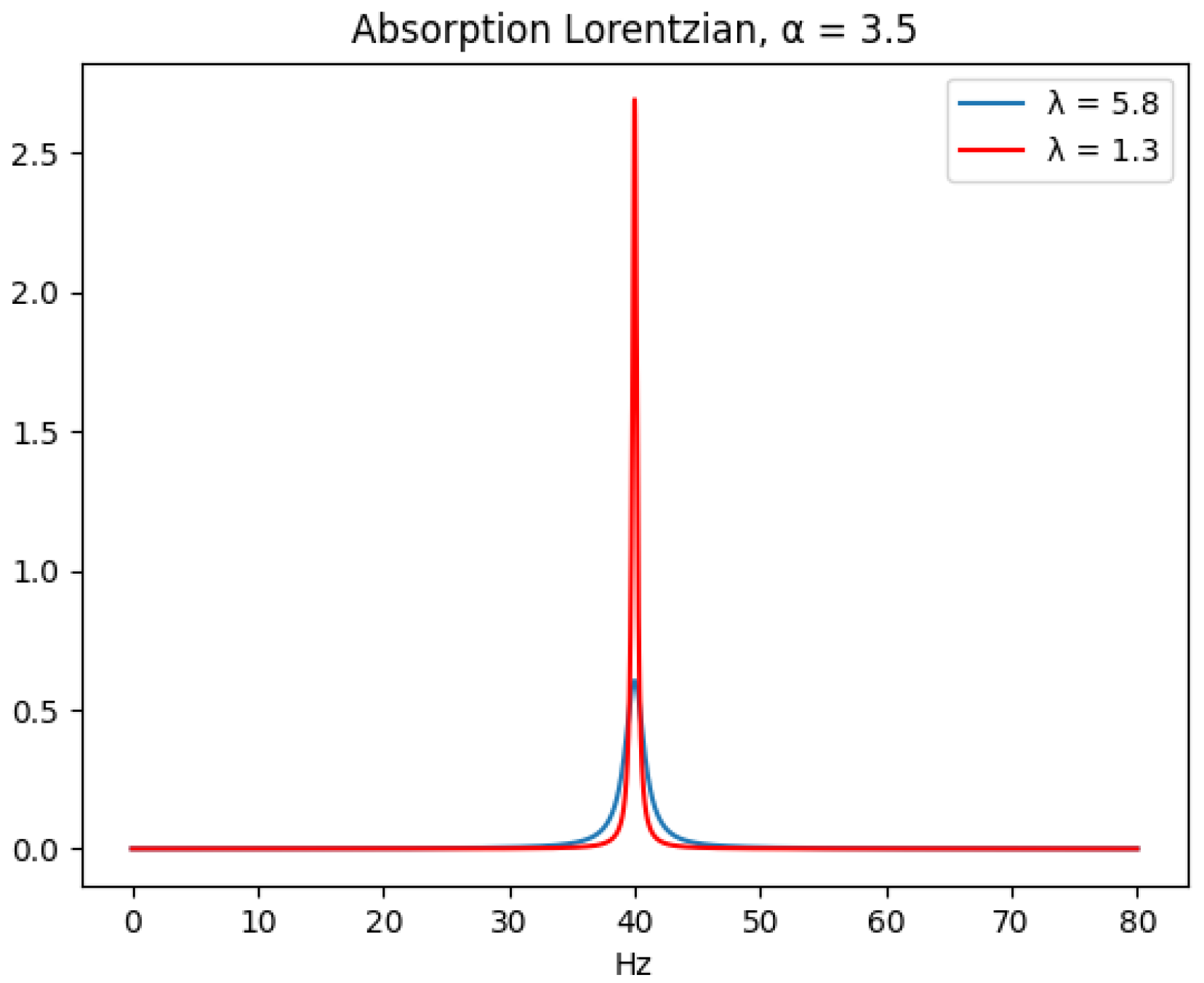
Existing quantifiers that use the FID signal model have the advantage that the FID model is a generative model for the data, i.e., no Fourier transform is used in the data model. This makes it possible to directly quantify uncertainty under a Bayesian inference framework. It does not have issues due to unpredictable baseline nor phase distortion that arise from Fourier transform approximation and autophasing. The disadvantage is that all of the FID parameters and the metabolite concentrations must be accurately estimated.
Working in the time domain involves fitting the model (Equation (
1)) to the observed time series (the FID data), but the overlapping set of an unknown number of resonance frequencies places significant computational strain on the numerical optimization algorithm used. Due to the number of FID variables involved with just a single metabolite, it is difficult to scale up this approach to handle the number of metabolites and experiments that are needed in a bioreactor monitoring application. Quantifiers that use the Fourier transformed FID signal model tend to require less computational resources, since the resonance frequencies are easier resolved in the Fourier domain without requiring accurate estimates of the other FID parameters. The quantifiers that we benchmark in
Section 5 all use this type of signal model.
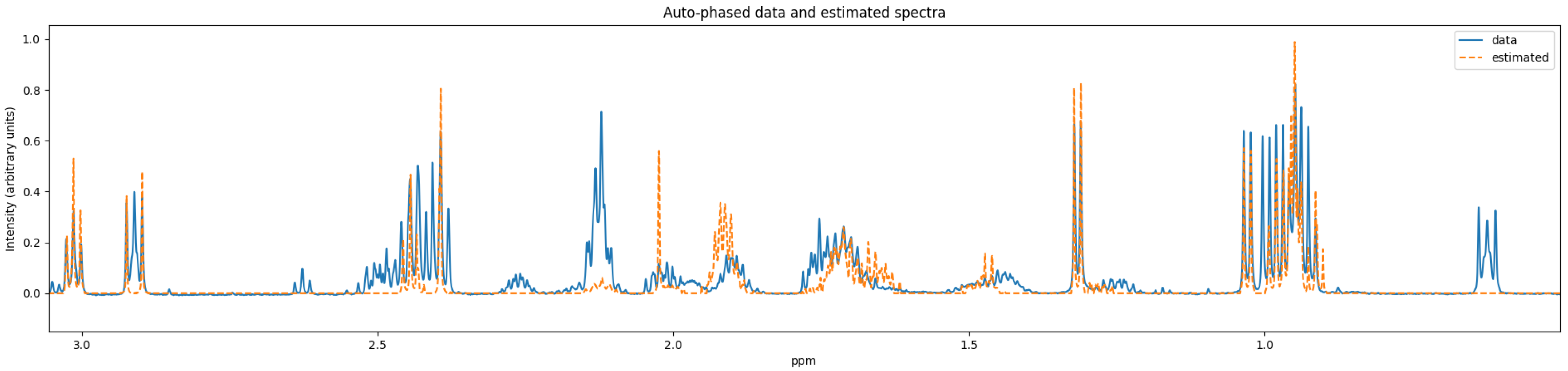
As discussed in
Section 3.6, most of the Fourier domain quantification schemes use some autophase algorithm as a preprocessing step to reduce the computational burden. However, these preprocessing steps could have issues when the signal-to-noise ratio of the NMR experiment is low. Since time-domain NMR quantification approaches typically avoid such preprocessing steps, they could perform better on experiments that are collected in low signal-to-noise ratio environments. Such a time-domain quantifier for quantifying a low number of compounds was reported by Matviychuk et al. [
42].
Some recent quantifiers place constraints on the FID parameters. E-RANSYS (Extractive ratio analysis NMR spectroscopy) [
43] and AQuA (Automated quantification algorithm) [
44] work by constraining the relative ratio of the amplitude of peaks of each metabolite in its internal library. ASICS (Automatic statistical identification in complex spectra) [
15] fits a deformation mapping of the peak positions to account for peak shifting. However, since the resonance frequency shiftdue to experimental conditions is hard to predict (see
Section 3.4), such a deformation mapping approach to predict the peak shifts should require a sizeable amount of data, sampled at various concentrations and chemical environment conditions. The promising report [
34] used a few thousand NMR experiments sampled at different concentrations to construct a predictive mapping for the chemical shift of selected metabolites that are common in urine. Peak alignment approaches such as [
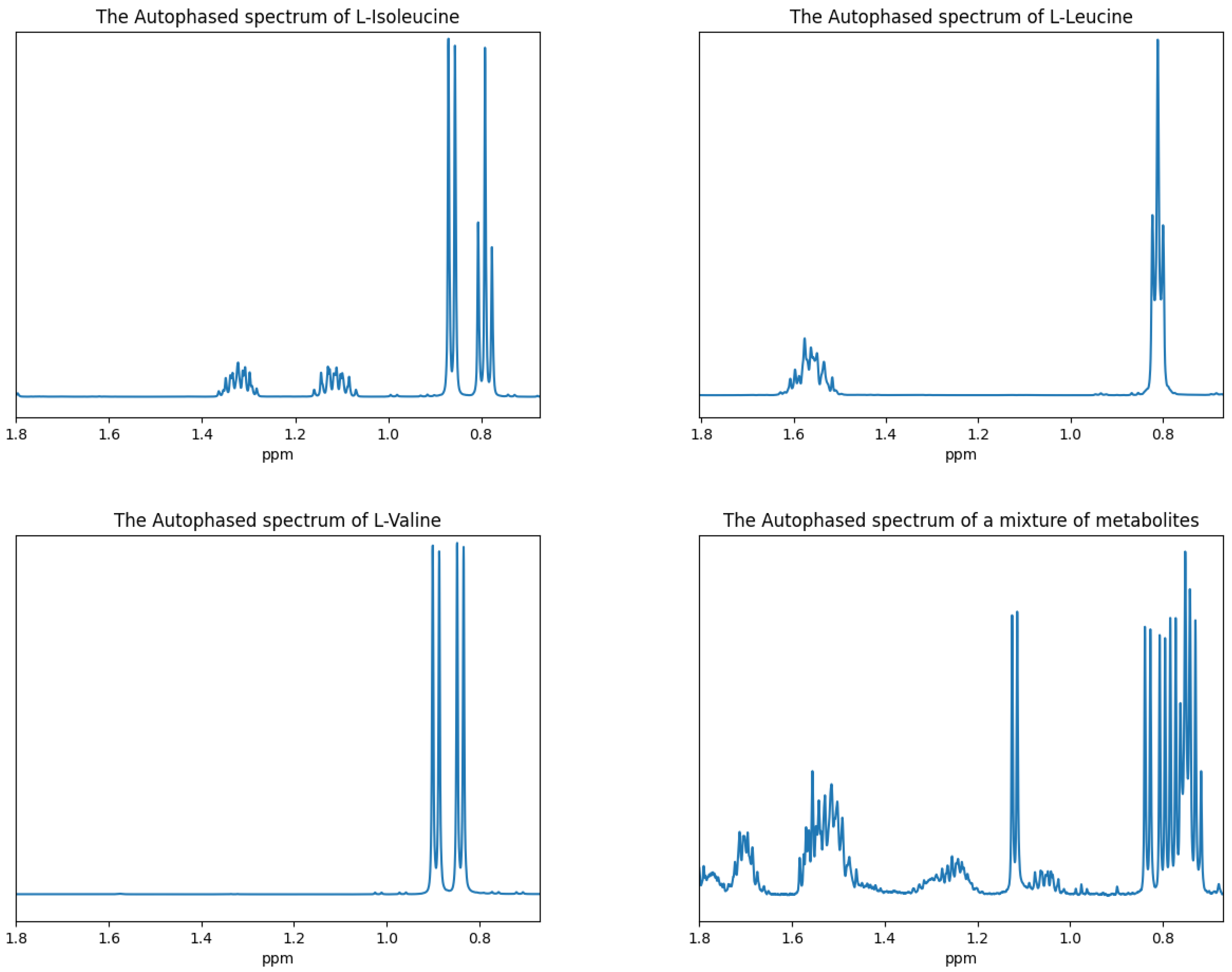
45] can be used prior to the quantification to compensate for peak shifts. Due to user-induced bias in the adjustment of peak positions, this approach can lead to assignment and overestimation errors.
Some quantifiers require input parameters that needs to be manually determined from the data of each NMR experiment to be quantified. The BATMAN (Bayesian automated metabolite analyzer for NMR) quantifier [
46] requires the user to specify various peak location and shift values of the targeted metabolites. This is a time-intensive and error-prone task, especially when the required information must be determined individually for each peak of each metabolite. Differences in spectral resolution of the instrument, peak overlap, and peak shift can lead to errors in this approach even after the extensive manual user-led input step.
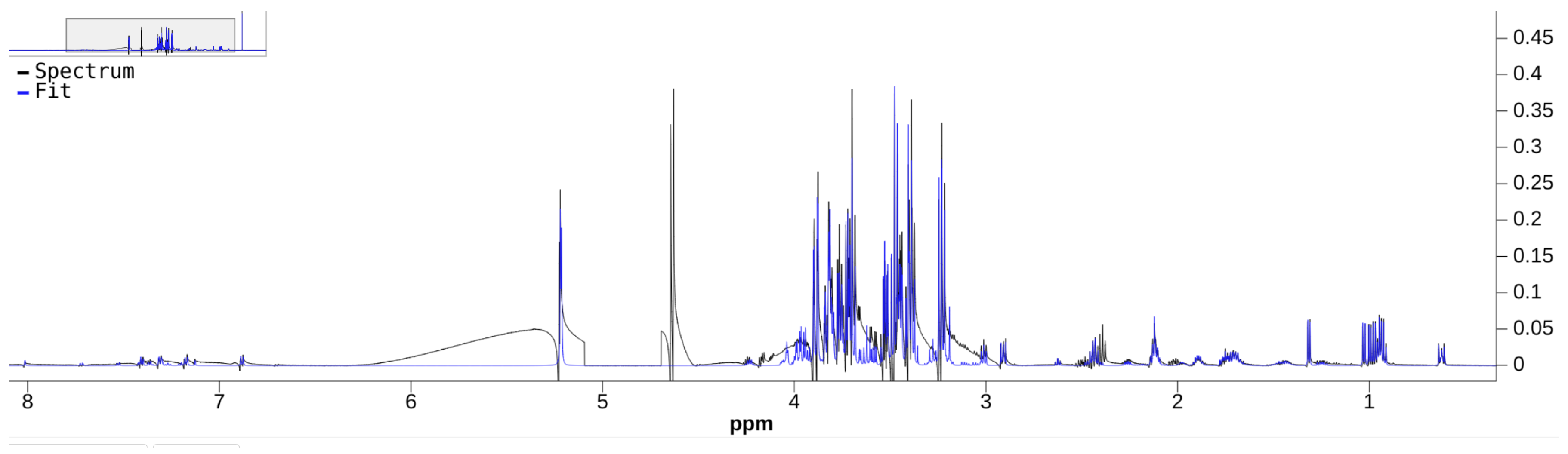
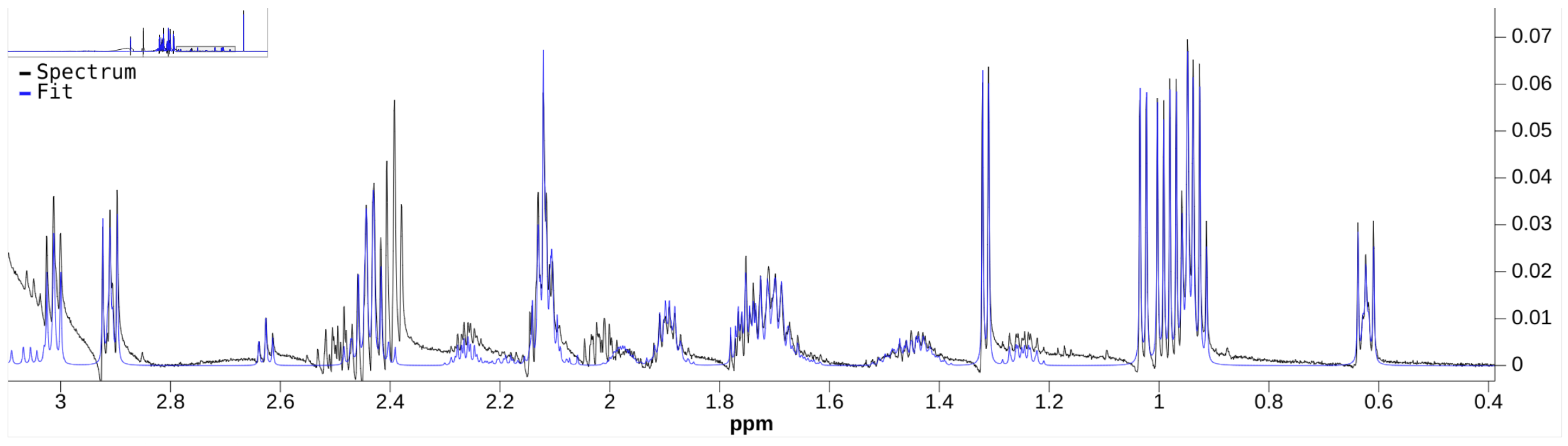
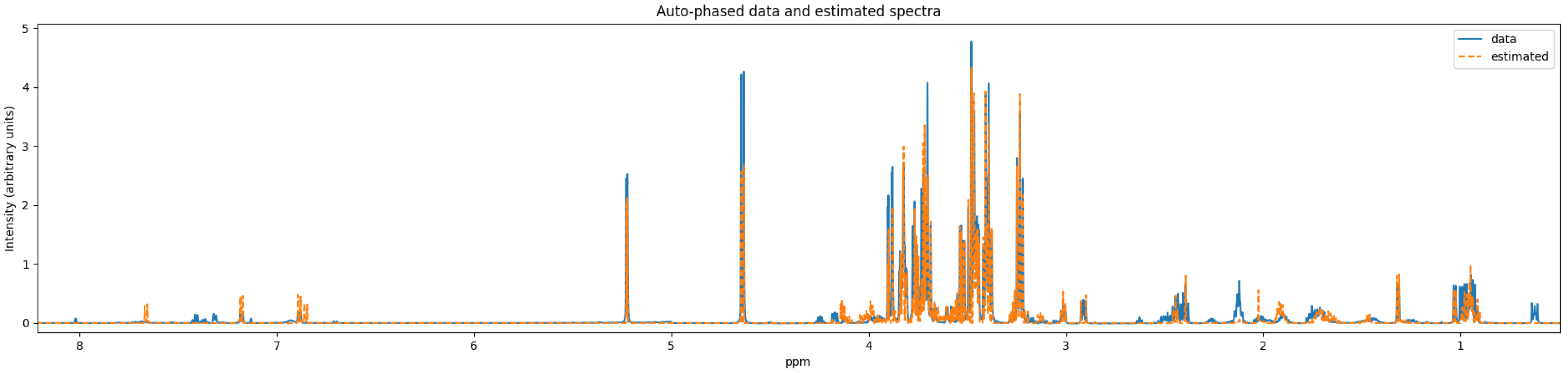
Although there are a few NMR quantifiers in the literature, some are no longer maintained, e.g., BATMAN. Many require proprietary software to run, e.g., E-RANSYS, AQuA, and the quantifier by Filntisi et al. [
47]. In this review, we focus on the Bayesil [
12], the ASICS [
15], and the rDolphin [
14] quantifiers. These quantifiers employ third-party autophasing and baseline correction as preprocessing steps, and are currently accessible to the public for running quantification jobs. They do not require extensive manual input of metabolite-specific instructions, making them appropriate for automated quantification.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}