
Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease




Abstract
1. Introduction
2. Metabolic Effects of Insulin
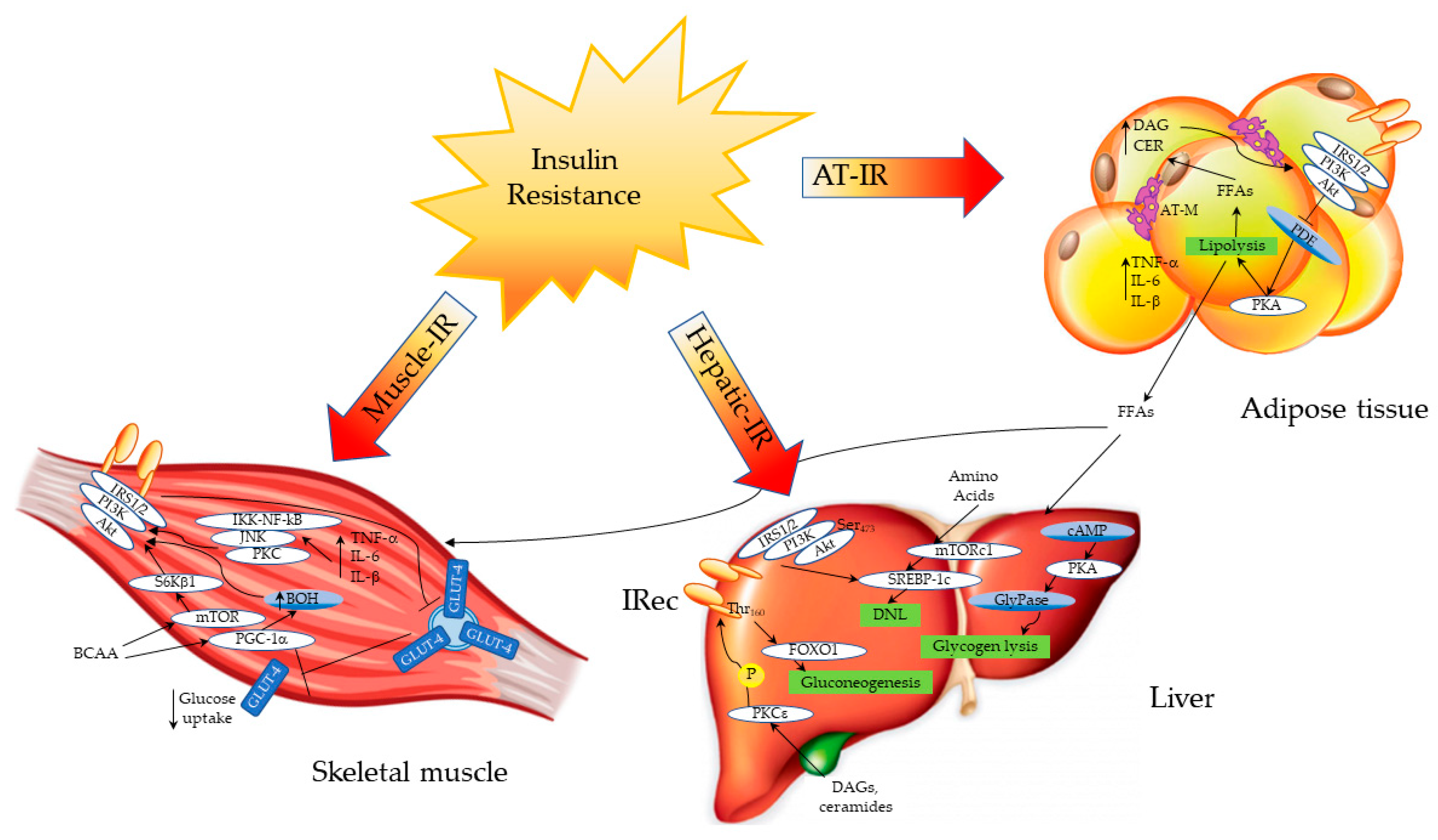
Sites and Mechanisms of Insulin Action and Insulin Resistance
3. Insulin Resistance in Nonalcoholic Fatty Liver Disease
3.1. Relation between Hepatic Steatosis and Insulin Resistance
3.2. Insulin Resistance in the Progression from Simple Steatosis to Nonalcoholic Steatohepatitis and Fibrosis
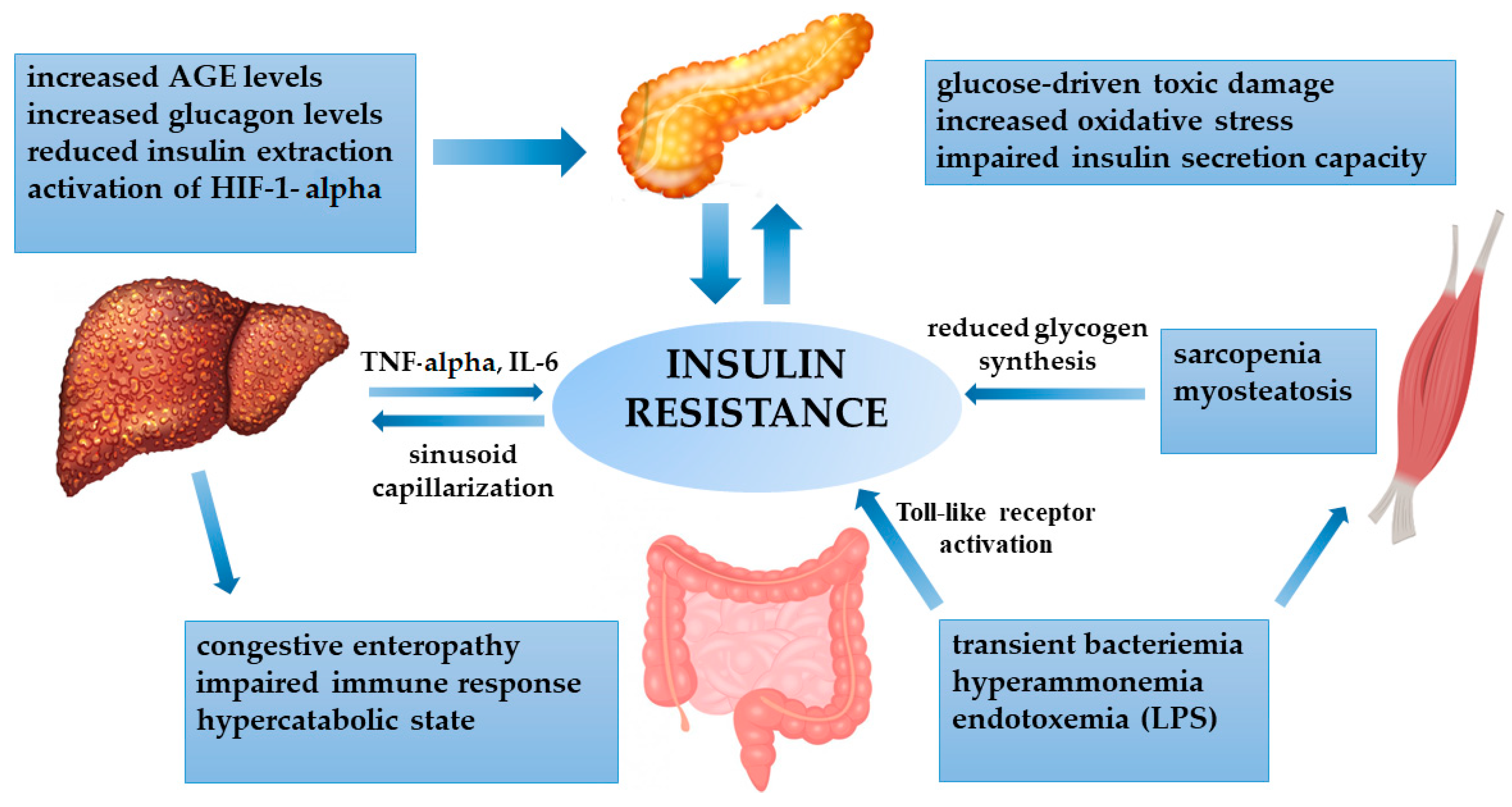
4. Insulin Resistance in Advanced Liver Disease
4.1. Insulin Resistance and Cirrhosis
4.2. Hepatogenous Diabetes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; McCollough, A.J.; Marchesini, G. Insulin resistance: A metabolic pathway to chronic liver disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- De Fronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, 157–163. [Google Scholar] [CrossRef]
- Shulman, G.I.; Rothman, D.L.; Jue, T.; Stein, P.; De Fronzo, R.A.; Shulman, R.G. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance. N. Engl. J. Med. 1990, 322, 223–228. [Google Scholar] [CrossRef]
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Brechtel-Hook, G.; Wallace, P.; Baron, A.D. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J. Clin. Investig. 1998, 101, 2377–2386. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; De Fronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 476279. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Muller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKC in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann. N. Y. Acad. Sci. 2002, 967, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Straczkowski, M.; Kowlaska, I.; Baranowski, M.; Nikolajuk, A.; Otziomek, E.; Zabielski, P.; Adamska, A.; Blachnio, A.; Gorski, J.; Gorska, M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia 2007, 50, 2366–2373. [Google Scholar] [CrossRef]
- Kolak, M.; Westerbacka, J.; Velagapudi, V.R.; Wågsäter, D.; Yetukuri, L.; Makkonen, J.; Rissanen, A.; Häkkinen, A.; Lindell, M.; Bergholm, R.; et al. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes 2007, 56, 1960–1968. [Google Scholar] [CrossRef]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Sadevirta, S.; Zhou, Y.; Kayser, B.; Ali, A.; Ahonen, L.; Lallukka, S.; Pelloux, V.; Gaggini, M.; Jian, C.; et al. Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugar. Diabetes Care 2018, 41, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Balage, M.; Dupont, J.; Mothe-Satney, I.; Tesseraud, S.; Mosoni, L.; Dardevet, D. Leucine supplementation in rats induced a delay in muscle IR/PI3K signalling pathway associated with overall impaired glucose tolerance. J. Nutr. Biochem. 2011, 22, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.R.; Liao, Y.; Hara, D.; Ding, Z.; Chen, C.; Nelson, J.L. An amino acid mixture improves glucose tolerance and insulin signalling in Sprague-Dawley rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, 752–760. [Google Scholar] [CrossRef]
- Zhang, Z.; Monleon, D.; Verhamme, P.; Staessen, J.A. Branched-chain amino acids as critical switches in health and disease. Hypertension 2018, 72, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.K.; Xu, X.J.; Lawson, E.; Deoliveira, R.; Brandon, A.E.; Kraegen, E.W.; Ruderman, N.B. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes 2010, 59, 2426–2434. [Google Scholar] [CrossRef] [PubMed]
- Lyon, E.S.; Rivera, M.E.; Johnson, M.A.; Sunderland, K.L.; Vaughan, R.A. Actions of chronic physiological 3-hydroxyisobuterate treatment on mitochondrial metabolism and insulin signalling in myotubes. Nutr. Res. 2019, 66, 22–31. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- De Fronzo, R.A.; Tobin, J.E.; Andres, R. Glucose clamp techniques: A method for quantifying insulin secretion and resistance. Am. J. Physiol. 1979, 237, 214–233. [Google Scholar] [CrossRef]
- Stephens, L.; Andreson, K.; Stokoe, D.; Erdjuoment-Bromage, H.; Painter, G.F.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; McCormick, F.; Tempst, P.; et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-triphosphate-dependent activation of protein kinase B. Science 1998, 279, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Swiderska, E.; Strycharz, J.; Wroblewski, A.; Szemraj, J.; Drzewoski, J.; Sliwinska, A. Role of PI3K/AKT Pathway in Insulin-Mediated Glucose Uptake; Open access peer-review chapter; Intechopen: London, UK, 2018. [Google Scholar] [CrossRef]
- Chandramouli, V.; Ekberg, K.; Schumann, W.C.; Kalhan, S.C.; Wahren, J.; Landau, B.R. Quantifying gluconeogenesis during fasting. Am. J. Physiol. 1997, 273, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Brown, M.S.; Goldstein, G.L. Bifurcation of insulin signalling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, I.; Rothman, D.L.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C NMR study. J. Clin. Investig. 1992, 90, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- O-Sullivan, I.S.; Zhang, W.; Wasserman, D.H.; Liew, C.W.; Liu, J.; Paik, J.; DePinho, R.A.; Stolz, D.B.; Kahn, C.R.; Schwartz, M.W.; et al. FOXO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef]
- Dong, X.C.; Copps, K.D.; Guo, S.; Li, Y.; Kollipara, R.; DePinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008, 8, 65–76. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Jeromson, S.; Levesque-Damphousse, P.; Secco, B.; Laplante, M.; Estall, J.L. PGC1a regulates the IRS1:IRS2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc. Nat. Acad. Sci. USA 2019, 116, 4285–4290. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Seale, J.P.; Donnelly, R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats—Effects of feeding. J. Endocrinol. 1999, 162, 207–214. [Google Scholar] [CrossRef]
- Akhtar, D.H.; Iqbal, U.; Vazquez-Montesino, L.M.; Dennis, B.B.; Ahmed, A. Pathogenesis of insulin resistance and atherogenic dyslipidemia in non-alcoholic fatty liver disease. J. Clin. Translat. Hepatol. 2019, 7, 362–370. [Google Scholar] [CrossRef]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanisms of insulin resistance in non-alcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S.; et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446. [Google Scholar] [CrossRef]
- Jaworski, K.; Sarkadi-Nagy, E.; Duncan, R.E.; Ahmadian, M.; Sul, H.S. Regulation of triglycerides metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between Adipocyte Size and Adipokine. Expression and Secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Jernas, M.; Palming, J.; Sjoholm, K.; Jennische, E.; Svensson, P.A.; Gabrielsson, B.G.; Levin, M.; Sjogren, A.; Rudemo, M.; Lystig, T.C.; et al. Separation of human adipocytes by size: Hypertrophic fat cells display distinct gene expression. FASEB J. 2006, 20, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Aguirre, V.; Kim, J.K.; Shulman, G.I.; Lee, A.; Corbould, A.; Dunaif, A.; White, M. Insulin/IGF-1 and TNF-a stimulate phosphorulation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Investig. 2001, 107, 181. [Google Scholar] [CrossRef]
- Da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Pagano, G.; Pacini, G.; Musso, G.; Gambino, R.; Mecca, F.; Depetris, N.; Cassader, M.; David, E.; Cavallo-Perin, P.; Rizzetto, M. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: Further evidence for an etiologic association. Hepatology 2002, 35, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Wong, G.L.; Yip, G.W.; Lo, A.O.; Limquiaco, J.; Chu, W.C.; Chim, A.M.; Yu, C.M.; Yu, J.; Chan, F.K.; et al. Coronary artery disease and cardiovascular outcomes in patients with non-alcoholic fatty liver disease. Gut 2011, 60, 1721–1727. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Bouter, L.M.; McQuillan, G.M.; Wener, M.H.; Harris, T.B. Elevated c-reactive protein levels in overweight and obese adults. J. Am. Med. Assoc. 1999, 282, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Vogelberg, K.H.; Gries, F.A.; Moschinski, D. Hepatic production of VLDL-triglycerides. Dependence of portal substrate and insulin concentration. Horm. Metab. Res. 1980, 12, 688–694. [Google Scholar] [CrossRef] [PubMed]
- McMilland, D.E. Increased levels of acute-phase serum proteins in diabetes. Metabolism 1989, 38, 1042–1046. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Qiang, L.; Kangsamaksin, T.; Kitajewski, J.; Ginsberg, H.N.; Accili, D. Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat. Med. 2013, 19, 1054–1060. [Google Scholar] [CrossRef]
- Rosso, C.; Kazankov, K.; Younes, R.; Esmaili, S.; Marietti, M.; Sacco, M.; Carli, F.; Salomone, F.; Gaggini, M.; Moller, H.J.; et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J. Hepatol. 2019, 71, 10112–11021. [Google Scholar] [CrossRef]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein-P, cause insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Rosso, C.; Armandi, A.; Gaggini, M.; Carli, F.; Abate, M.L.; Olivero, A.; Ribaldone, D.G.; Saracco, G.M.; Gastaldelli, A.; et al. Interplay between oxidative stress and metabolic derangements in non-alcoholic fatty liver disease: The role of selenoprotein P. Int. J. Mol. Sci. 2020, 21, 8838. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Navari, N.; Vivoli, E.; Galastri, S.; Provenzano, A. Modulation of liver fibrosis by adipokines. Dig. Dis. 2011, 29, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Bertolani, C.; Marra, F. Role of adipocytokines in hepatic fibrosis. Curr. Pharm. Des. 2010, 16, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A. Insulin-like growth factor 1 and non-alcoholic fatty liver disease: A systemic review and meta-analysis. Endocrine 2019, 65, 227–237. [Google Scholar] [CrossRef]
- Yao, Y.; Miao, X.; Zhu, D.; Li, D.; Zhang, Y.; Song, C.; Liu, K. Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis via suppressors of cytokine signaling (SOCS-3). J. Cell. Biochem. 2010, 110, 1195–1207. [Google Scholar] [CrossRef]
- Hagstrom, H.; Stal, P.; Hultcrantz, R.; Brismar, K.; Ansurudeen, I. IGFBP-1 and IGF-1 as markers for advanced fibrosis in NAFLD—A pilot study. Scand. J. Gastroenterol. 2017, 52, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Jiang, X.; Li, J.; Bai, Y.; Li, Z.; Wei, P.; Sun, S.; Liang, Y.; Han, S.; Li, X.; et al. Insulin-like growth factor-1 attenuates oxidative stress-induced hepatocyte premature senescence in liver fibrogenesis via regulating nuclear p53–progerin interaction. Cell Death Dis. 2019, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Ann. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Handy, J.A.; Fu, P.P.; Kumar, P.; Mells, J.E.; Sharma, S.; Saxena, N.K.; Anania, F.A. Adiponectin inhibits leptin signalling via multiple mechanisms to exert protective effects against hepatic fibrosis. Biochem. J. 2011, 440, 385–395. [Google Scholar] [CrossRef]
- Zhao, L.; Fu, Z.; Liu, Z. Adiponectin and insulin crosstalk: The microvascular connection. Trends Cardiovasc. Med. 2014, 24, 319–324. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Da Silva Morais, A.; Schroyen, B.; Van Hul, N.; Geerts, A. Insulin resistance in hepatocytes and sinusoidal liver cells: Mechanisms and consequences. J. Hepatol. 2007, 47, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to non-alcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Krawczyk, M.; Stokes, C.S.; Romeo, S.; Lammert, F. HCC and liver disease risks in homozygous PNPLA3 p.I148M carriers approach monogenic inheritance. J. Hepatol. 2015, 62, 980–981. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.J.; Willmann, O.; Rieger, A.; Fenk, A.; Selberg, O.; Lautz, H.U.; Bürger, M.; Balks, H.J.; von zur Mühlen, A.; Schmidt, F.W. Mechanism of insulin resistance associated with liver cirrhosis. Gastroenterology 1992, 102, 2033–2041. [Google Scholar] [CrossRef]
- Moscatiello, S.; Manini, R.; Marchesini, G. Diabetes and liver disease: An ominous association. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 63–70. [Google Scholar] [CrossRef]
- Goral, V.; Atalay, R.; Kucukoner, M. Insulin resistance in liver cirrhosis. Hepatogastroenterology 2010, 57, 309–315. [Google Scholar]
- Goswami, A.; Bhargava, N.; Dadhich, S.; Kulamarva, G. Insulin resistance in euglycemic cirrhosis. Ann. Gastroenterol. 2014, 27, 237–243. [Google Scholar]
- Misu, H. Identification of hepatokines involved in pathology of type 2 diabetes and obesity. Endocr. J. 2019, 29, 659–662. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Phillips, I.D.; Arany, E.; Strain, A.J.; Han, V.K.; Hill, D.J. Rapid clearance of insulin-like growth factor (IGF)-binding protein species from blood and an associated fall in circulating IGF-I following partial hepatectomy in the rat. J. Endocrinol. 1993, 137, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Shiratsuki, S.; Matsuda, T.; Iwamoto, T.; Takami, T.; Uchida, K.; Terai, S.; Yamasaki, T.; Sakaida, I. Occlusion of portosystemic shunts improves hyperinsulinemia due to insulin resistance in cirrhotic patients with portal hypertension. J. Gastroenterol. 2014, 49, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Bhanji, R.A.; Mazurak, V.C.; Montano-Loza, A.J. Sarcopenia in cirrhosis: From pathogenesis to interventions. J. Gastroenterol. 2019, 54, 845–859. [Google Scholar] [CrossRef]
- Bhanji, R.A.; Moctezuma-Velazquez, C.; Duarte-Rojo, A.; Ebadi, M.; Ghosh, S.; Rose, C.; Montano-Loza, A.J. Myosteatosis and sarcopenia are associated with hepatic encephalopathy in patients with cirrhosis. Hepatol. Int. 2018, 12, 377–386. [Google Scholar] [CrossRef]
- Lanthier, N.; Molendi-Coste, O.; Cani, P.D.; van Rooijen, N.; Horsmans, Y.; Leclercq, I.A. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J. 2011, 25, 4301–4311. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Navasa, M.; Gómez, J.; Colmenero, J.; Vila, J.; Arroyo, V.; Rodés, J. Bacterial infections in cirrhosis: Epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology 2002, 35, 140–148. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Kruszynska, Y.T.; Goulas, S.; Wollen, N.; McIntyre, N. Insulin secretory capacity and the regulation of glucagon secretion in diabetic and non-diabetic alcoholic cirrhotic patients. J. Hepatol. 1998, 28, 280–291. [Google Scholar] [CrossRef]
- Petrides, A.S.; Vogt, C.; Schulze-Berge, D.; Matthews, D.; Strohmeyer, G. Pathogenesis of glucose intolerance and diabetes mellitus in cirrhosis. Hepatology 1994, 19, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Picardi, A.; D’Avola, D.; Gentilucci, U.V.; Galati, G.; Fiori, E.; Spataro, S.; Afeltra, A. Diabetes in chronic liver disease: From old concepts to new evidence. Diabetes Metab. Res. Rev. 2006, 22, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Makita, Z.; Horii, Y.; Brunelle, S.; Cerami, A.; Sehajpal, P.; Suthanthiran, M.; Vlassara, H. Two novel rat liver membrane proteins that bind advanced glycosylation endproducts: Relationship to macrophage receptor for glucose-modified proteins. J. Exp. Med. 1991, 174, 515–524. [Google Scholar] [CrossRef]
- Moreau, R.; Lee, S.S.; Soupison, T.; Roche-Sicot, J.; Sicot, C. Abnormal tissue oxygenation in patients with cirrhosis and liver failure. J. Hepatol. 1988, 7, 98–105. [Google Scholar] [CrossRef]
- Cheng, K.; Ho, K.; Stokes, R.; Scott, C.; Lau, S.M.; Hawthorne, W.J.; O’Connell, P.J.; Loudovaris, T.; Kay, T.W.; Kulkarni, R.N.; et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J. Clin. Investig. 2010, 120, 2171–2183. [Google Scholar] [CrossRef]
- García-Compeán, D.; Jáquez-Quintana, J.O.; Lavalle-González, F.J.; Reyes-Cabello, E.; González-González, J.A.; Muñoz-Espinosa, L.E.; Vázquez-Elizondo, G.; Villarreal-Pérez, J.Z.; Maldonado-Garza, H.J. The prevalence and clinical characteristics of glucose metabolism disorders in patients with liver cirrhosis. A prospective study. Ann. Hepatol. 2012, 11, 240–248. [Google Scholar] [CrossRef]
- Orsi, E.; Grancini, V.; Menini, S.; Aghemo, A.; Pugliese, G. Hepatogenous diabetes: Is it time to separate it from type 2 diabetes? Liver Int. 2017, 37, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Holstein, H.; Hinze, S.; Thiessen, E.; Plaschke, A.; Egberts, E.H. Clinical implications of hepatogenous diabetes in liver cirrhosis. J. Gastroenterol. Hepatol. 2002, 17, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Gentile, S.; Loguercio, C.; Marmo, R.; Carbone, L.; Del Vecchio Blanco, C. Incidence of altered glucose tolerance in liver cirrhosis. Diabetes Res. Clin. Pract. 1993, 22, 37–44. [Google Scholar] [CrossRef]
- Perseghin, G.; Mazzaferro, V.; Benedini, S.; Pulvirenti, A.; Coppa, J.; Regalia, E.; Luzi, L. Resting energy expenditure in diabetic and nondiabetic patients with liver cirrhosis: Relation with insulin sensitivity and effect of liver transplantation and immunosuppressive therapy. Am. J. Clin. Nutr. 2002, 76, 541–548. [Google Scholar] [CrossRef] [PubMed]
- García-Compeán, D.; González-González, J.A.; Lavalle-González, F.J.; González-Moreno, E.I.; Villarreal-Pérez, J.Z.; Maldonado-Garza, H.J. Hepatogenous diabetes: Is it a neglected condition in chronic liver disease? World J. Gastroenterol. 2016, 22, 2869–2874. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Taniguchi, E.; Itou, M.; Sakata, M.; Sumie, S.; Sata, M. Insulin resistance and chronic liver disease. World J. Hepatol. 2011, 3, 99–107. [Google Scholar] [CrossRef]
- Trenti, T.; Cristani, A.; Cioni, G.; Pentore, R.; Mussini, C.; Ventura, E. Fructosamine and glycated hemoglobin as indices of glycemic control in patients with liver cirrhosis. Ric. Clin. Lab. 1990, 20, 261–267. [Google Scholar] [CrossRef]
- Proietto, J.; Alford, F.P.; Dudley, F.J. The mechanism of the carbohydrate intolerance of cirrhosis. J. Clin. Endocrinol. Metab. 1980, 51, 1030–1036. [Google Scholar] [CrossRef]
- Petrides, A.S.; Groop, L.C.; Riely, C.A.; DeFronzo, R.A. Effect of physiologic hyperinsulinemia on glucose and lipid metabolism in cirrhosis. J. Clin. Investig. 1991, 88, 561–570. [Google Scholar] [CrossRef]
- Marselli, L.; De Simone, P.; Morganti, R.; Coletti, L.; Carrai, P.; Catalano, G.; Tincani, G.; Ghinolfi, D.; Occhipinti, M.; Filipponi, F.; et al. Frequency and characteristics of diabetes in 300 pre-liver transplant patients. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 441–442. [Google Scholar] [CrossRef]
- Bianchi, G.; Marchesini, G.; Zoli, M.; Bugianesi, E.; Fabbri, A.; Pisi, E. Prognostic significance of diabetes in patients with cirrhosis. Hepatology 1994, 20, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Moreau, R.; Delègue, P.; Pessione, F.; Hillaire, S.; Durand, F.; Lebrec, D.; Valla, D.C. Clinical characteristics and outcome of patients with cirrhosis and refractory ascites. Liver Int. 2004, 24, 457–464. [Google Scholar] [CrossRef]
- Quintana, J.O.; García-Compean, D.; González, J.A.; Pérez, J.Z.; González, F.J.; Espinosa, L.E.; Hernández, P.L.; Cabello, E.R.; Villarreal, E.R.; Rendón, R.F.; et al. The impact of diabetes mellitus in mortality of patients with compensated liver cirrhosis—A prospective study. Ann. Hepatol. 2011, 10, 56–62. [Google Scholar] [CrossRef]
- Sangiovanni, A.; Prati, G.M.; Fasani, P.; Ronchi, G.; Romeo, R.; Manini, M.; Del Ninno, E.; Morabito, A.; Colombo, M. The natural history of compensated cirrhosis due to hepatitis C virus: A 17-year cohort study of 214 patients. Hepatology 2006, 43, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Berman, K.; Tandra, S.; Forssell, K.; Vuppalanchi, R.; Burton, J.R., Jr.; Nguyen, J.; Mullis, D.; Kwo, P.; Chalasani, N. Incidence and predictors of 30-day readmission among patients hospitalized for advanced liver disease. Clin. Gastroenterol. Hepatol. 2011, 3, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Elkrief, L.; Chouinard, P.; Bendersky, N.; Hajage, D.; Larroque, B.; Babany, G.; Kutala, B.; Francoz, C.; Boyer, N.; Moreau, R.; et al. Diabetes mellitus is an independent prognostic factor for major liver-related outcomes in patients with cirrhosis and chronic hepatitis C. Hepatology 2014, 60, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Sigal, S.H.; Stanca, C.M.; Kontorinis, N.; Bodian, C.; Ryan, E. Diabetes mellitus is associated with hepatic encephalopathy in patients with HCV cirrhosis. Am. J. Gastroenterol. 2006, 101, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Elkrief, L.; Rautou, P.E.; Sarin, S.; Valla, D.; Paradis, V.; Moreau, R. Diabetes mellitus in patients with cirrhosis: Clinical implications and management. Liver Int. 2016, 36, 936–948. [Google Scholar] [CrossRef]
- Gentile, S.; Guarino, G.; Romano, M.; Alagia, I.A.; Fierro, M.; Annunziata, S.; Magliano, P.L.; Gravina, A.G.; Torella, R. A randomized controlled trial of acarbose in hepatic encephalopathy. Clin. Gastroenterol. Hepatol. 2005, 3, 184–191. [Google Scholar] [CrossRef]
- Trail, K.C.; Stratta, R.J.; Larsen, J.L.; Ruby, E.I.; Patil, K.D.; Langnas, A.N.; Donovan, J.P.; Sorrell, M.F.; Zetterman, R.K.; Pillen, T.J.; et al. Results of liver transplantation in diabetic recipients. Surgery 1993, 114, 650–656. [Google Scholar]
- Yang, W.S.; Va, P.; Bray, F.; Gao, S.; Gao, J.; Li, H.L.; Xiang, Y.B. The role of pre-existing diabetes mellitus on hepatocellular carcinoma occurrence and prognosis: A meta-analysis of prospective cohort studies. PLoS ONE 2011, 6, e27326. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Ann. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Honda, M.; Asonuma, K.; Hayashida, S.; Suda, H.; Ohya, Y.; Lee, K.J.; Yamamoto, H.; Takeichi, T.; Inomata, Y. Incidence and risk factors for new-onset diabetes in living-donor liver transplant recipients. Clin. Transplant. 2013, 27, 426–435. [Google Scholar] [CrossRef]
- Chakkera, H.A.; Mandarino, L.J. Calcineurin inhibition and new-onset diabetes mellitus after transplantation. Transplantation 2013, 95, 647–652. [Google Scholar] [CrossRef]
- Lunati, M.E.; Grancini, V.; Agnelli, F.; Gatti, S.; Masserini, B.; Zimbalatti, D.; Pugliese, G.; Rossi, G.; Donato, M.F.; Colombo, M.; et al. Metabolic syndrome after liver transplantation: Short-term prevalence and pre- and post-operative risk factors. Dig. Liver Dis. 2013, 45, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G.; Mazzaferro, V.; Sereni, L.P.; Regalia, E.; Benedini, S.; Bazzigaluppi, E.; Pulvirenti, A.; Leão, A.A.; Calori, G.; Romito, R.; et al. Contribution of reduced insulin sensitivity and secretion to the pathogenesis of hepatogenous diabetes: Effect of liver transplantation. Hepatology 2000, 31, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Tsuji, S.; Tsujii, M.; Arimitsu, S.; Haruna, Y.; Imano, E.; Suzuki, M.; Kanda, T.; Kawano, S.; Hiramatsu, N.; et al. Oral glucose tolerance test predicts prognosis of patients with liver cirrhosis. Am. J. Gastroenterol. 2006, 101, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Grancini, V.; Trombetta, M.; Lunati, M.E.; Zimbalatti, D.; Boselli, M.L.; Gatti, S.; Donato, M.F.; Resi, V.; D’Ambrosio, R.; Aghemo, A.; et al. Contribution of beta-cell dysfunction and insulin resistance to cirrhosis-associated diabetes: Role of severity of liver disease. J. Hepatol. 2015, 63, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Grancini, V.; Trombetta, M.; Lunati, M.E.; Boselli, M.L.; Gatti, S.; Donato, M.F.; Palmieri, E.; Resi, V.; Pugliese, G.; Bonadonna, R.C.; et al. Central role of the beta-cell in driving regression of diabetes after liver transplantation in cirrhotic patients. J. Hepatol. 2019, 70, 954–962. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Year of Publication | Number of Patients | Follow-Up (Months) | Outcome |
|---|---|---|---|---|
| Bianchi et al. [98] | 1984 | 382 * | 37 | death (HR = 2.30, p = 0.019) |
| Moreau et al. [99] | 2004 | 75 * | 18 | death (HR = 2.20, p = 0.03) |
| Sangiovanni et al. [101] | 2006 | 214 | 204 | not significant |
| Berman et al. [102] | 2011 | 447 * | 3 | not significant |
| Quintana et al. [100] | 2011 | 110 | 30 | death (OR = 3.30, p = 0.007) ** |
| Elkrief et al. [103] | 2014 | 348 * | 60 | ascites (OR = 1.70, p = 0.05) |
| bacterial infections (OR = 3.02, p = 0.001) | ||||
| HE (OR = 6.55, p < 0.001) | ||||
| HCC (OR = 1.93, p = 0.016) | ||||
| death (HR = 1.33, p = 0.027) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 155. https://doi.org/10.3390/metabo11030155
Armandi A, Rosso C, Caviglia GP, Bugianesi E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites. 2021; 11(3):155. https://doi.org/10.3390/metabo11030155
Chicago/Turabian StyleArmandi, Angelo, Chiara Rosso, Gian Paolo Caviglia, and Elisabetta Bugianesi. 2021. "Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease" Metabolites 11, no. 3: 155. https://doi.org/10.3390/metabo11030155
APA StyleArmandi, A., Rosso, C., Caviglia, G. P., & Bugianesi, E. (2021). Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites, 11(3), 155. https://doi.org/10.3390/metabo11030155