
Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry

 , ,
, ,
Abstract
1. Introduction
2. Results
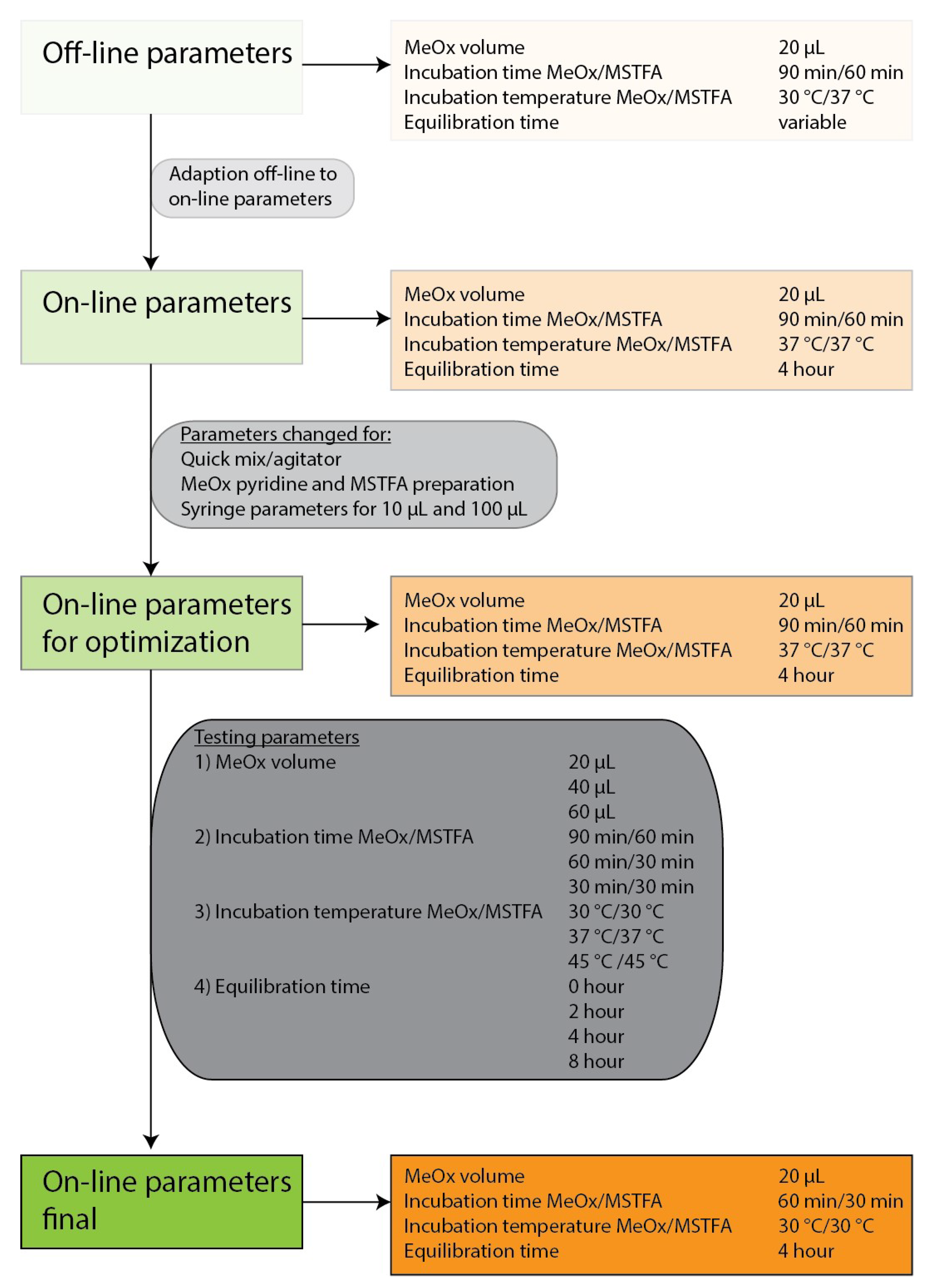
2.1. Optimization of the Derivatization Parameters
2.1.1. MeOx Volume
2.1.2. Incubation Time
2.1.3. Incubation Temperature
2.1.4. Equilibration Time
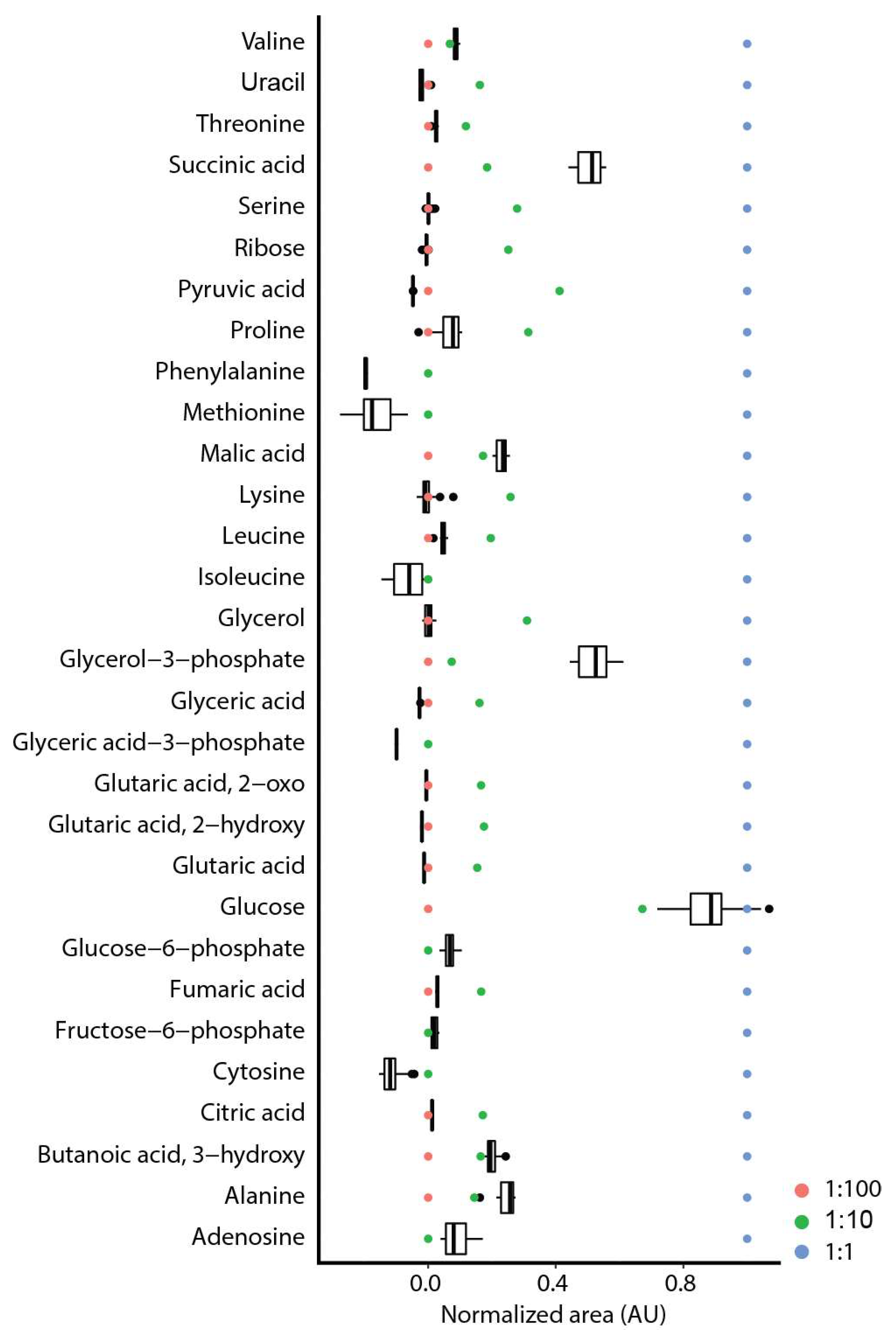
2.2. Repeatability and Reproducibility
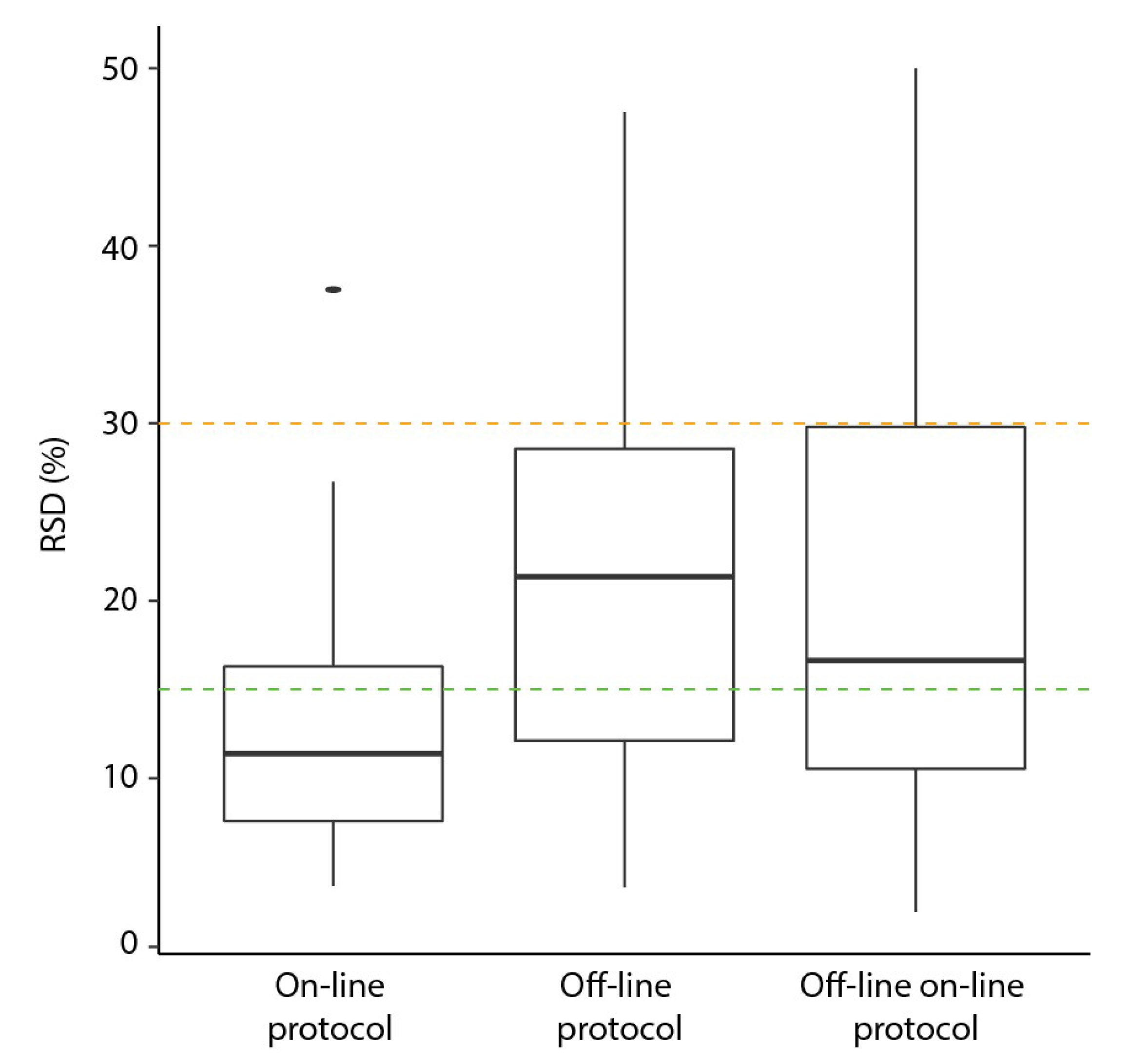
2.3. Comparison of on-Line to off-Line Derivatization
3. Discussion
4. Materials and Methods
4.1. Extraction of Calibration Standards
4.2. Plasma and Serum Extraction
4.3. Liver Extraction
4.4. GC-MS Metabolomics Measurement of Key Central Carbon Pathway Metabolites
4.4.1. On-line Derivatization
4.4.2. Off-line Derivatization
4.4.3. Instrumentation
4.4.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halket, J.M.; Waterman, D.; Przyborowska, A.M.; Patel, R.K.; Fraser, P.D.; Bramley, P.M. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J. Exp. Bot. 2005, 56, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Villas-Boas, S.G.; Smart, K.F.; Sivakumaran, S.; Lane, G.A. Alkylation or Silylation for Analysis of Amino and Non-Amino Organic Acids by GC-MS? Metabolites 2011, 1, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; David, F.; Lynen, F.; Rumpel, K.; Dugardeyn, J.; Van Der Straeten, D.; Xu, G.; Sandra, P. Evaluation of automated sample preparation, retention time locked gas chromatography-mass spectrometry and data analysis methods for the metabolomic study of Arabidopsis species. J. Chromatogr. A 2011, 1218, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Abbiss, H.; Rawlinson, C.; Maker, G.L.; Trengove, R. Assessment of automated trimethylsilyl derivatization protocols for GC–MS-based untargeted metabolomic analysis of urine. Metabolomics 2015, 11, 1908–1921. [Google Scholar] [CrossRef]
- Fritsche-Guenther, R.; Gloaguen, Y.; Kirchner, M.; Mertins, P.; Tunn, P.U.; Kirwan, J.A. Progression-Dependent Altered Metabolism in Osteosarcoma Resulting in Different Nutrient Source Dependencies. Cancers 2020, 12, 1371. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Blaschke, B.; Benn, A.; Hammer, E.; Witt, E.; Kirwan, J.; Fritsche-Guenther, R.; Gloaguen, Y.; Bartsch, C.; Vietzke, A.; et al. Sex-specific metabolic and functional differences in human umbilical vein endothelial cells from twin pairs. Atherosclerosis 2019, 291, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Fritsche-Guenther, R.; Zasada, C.; Mastrobuoni, G.; Royla, N.; Rainer, R.; Rossner, F.; Pietzke, M.; Klipp, E.; Sers, C.; Kempa, S. Alterations of mTOR signaling impact metabolic stress resistance in colorectal carcinomas with BRAF and KRAS mutations. Sci. Rep. 2018, 8, 9204. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics 2011, 7, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Shi, S.; Yi, Z.; He, R.; Lu, H.; Liang, Y. MeOx-TMS derivatization for GC-MS metabolicprofiling of urine and application in thediscrimination between normal C57BL/6J and type2 diabetic KK-Ay mice. AnalyticalMethods 2014, 6, 4380. [Google Scholar] [CrossRef]
- Gullberg, J.; Jonsson, P.; Nordstrom, A.; Sjostrom, M.; Moritz, T. Design of experiments: An efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 2004, 331, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Pietzke, M.; Zasada, C.; Mudrich, S.; Kempa, S. Decoding the dynamics of cellular metabolism and the action of 3-bromopyruvate and 2-deoxyglucose using pulsed stable isotope-resolved metabolomics. Cancer Metab. 2014, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Opialla, T.; Kempa, S.; Pietzke, M. Towards a More Reliable Identification of Isomeric Metabolites Using Pattern Guided Retention Validation. Metabolites 2020, 10, 457. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter- | MeOx Volume | Time | Temperature | Equilibration |
|---|---|---|---|---|
| Analyzed replicates | 20 µL: 5 40 µL: 5 60 µL: 4 | 30/30 min: 4 60/30 min: 3 90/60 min: 5 | 30 °C: 4 37 °C: 3 45 °C: 3 | 0 h: 4 2 h: 4 4 h: 5 8 h: 4 |
| Detected compounds | 20 µL: 32 40 µL: 33 60 µL: 32 | 30/30 min: 33 60/30 min: 34 90/60 min: 31 | 30 °C: 38 37 °C: 37 45 °C: 36 | 0 h: 34 2 h: 36 4 h: 36 8 h: 34 |
| Median RSD (%) | 20 µL: 17 40 µL: 27 60 µL: 33 | 30/30 min: 23 60/30 min: 14 90/60 min: 18 | 30 °C: 10 37 °C: 10 45 °C: 21 | 0 h: 11 2 h: 21 4 h: 15 8 h: 15 |
| Parameter | Plasma | Liver | Batch 1 | Batch 2 | Batch 3 |
|---|---|---|---|---|---|
| Number of metabolites | 0.5 | 0 | 1.9 | 2.4 | 2.2 |
| Number of missing values | 11 (0.8%) | 0 (0%) | 24 (11%) | 25 (12%) | 25 (12%) |
| Median RSD | 16% | 10% | 21% | 20% | 19% |
| RSD range | 11–28% | 2–56% | 3–42% | 12–69% | 13–39% |
| Parameter | On-Line | Off-Line (Original) | Off-Line with On-Line Settings (OLOLP) |
|---|---|---|---|
| Replicates | 9 | 9 | 8 |
| Number metabolites | 0.73 | 0.83 | 0.71 |
| Number of missing values | 14 (6%) | 7 (3%) | 5 (3%) |
| Median RSD | 11% | 21% | 17% |
| RSD range | 4–38% | 4–48% | 2–50% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritsche-Guenther, R.; Gloaguen, Y.; Bauer, A.; Opialla, T.; Kempa, S.; Fleming, C.A.; Redmond, H.P.; Kirwan, J.A. Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry. Metabolites 2021, 11, 888. https://doi.org/10.3390/metabo11120888
Fritsche-Guenther R, Gloaguen Y, Bauer A, Opialla T, Kempa S, Fleming CA, Redmond HP, Kirwan JA. Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry. Metabolites. 2021; 11(12):888. https://doi.org/10.3390/metabo11120888
Chicago/Turabian StyleFritsche-Guenther, Raphaela, Yoann Gloaguen, Anna Bauer, Tobias Opialla, Stefan Kempa, Christina A. Fleming, Henry Paul Redmond, and Jennifer A. Kirwan. 2021. "Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry" Metabolites 11, no. 12: 888. https://doi.org/10.3390/metabo11120888
APA StyleFritsche-Guenther, R., Gloaguen, Y., Bauer, A., Opialla, T., Kempa, S., Fleming, C. A., Redmond, H. P., & Kirwan, J. A. (2021). Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry. Metabolites, 11(12), 888. https://doi.org/10.3390/metabo11120888