
The Reciprocal Relationship between LDL Metabolism and Type 2 Diabetes Mellitus

 ,
,  , ,
, ,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Low-Density Lipoproteins
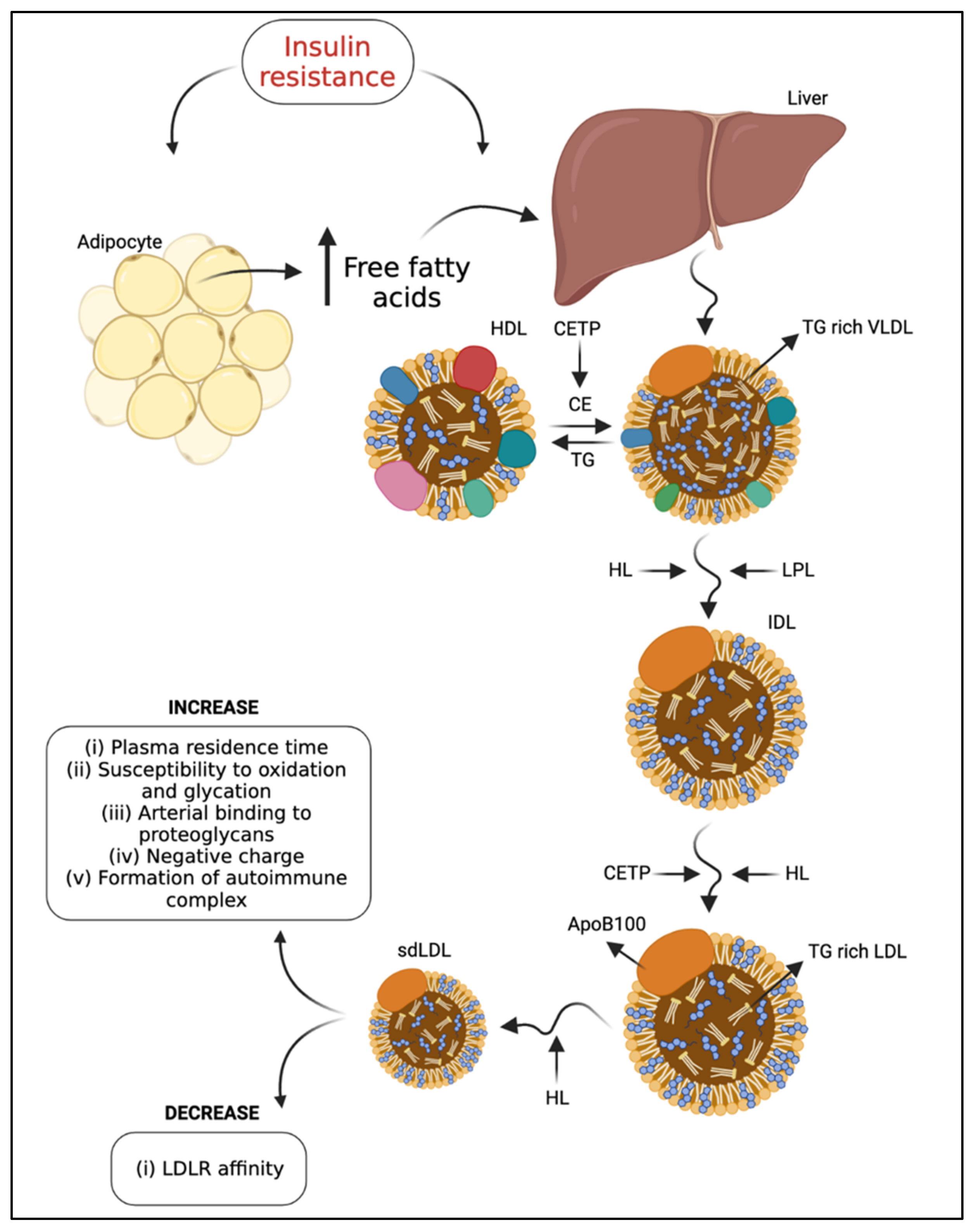
3. Influences on LDL Subfraction Heterogeneity
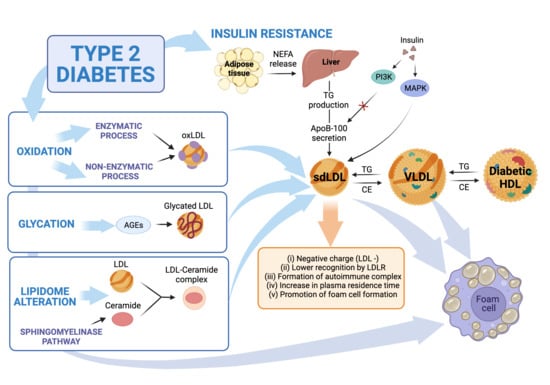
4. LDL Modification Due to T2DM
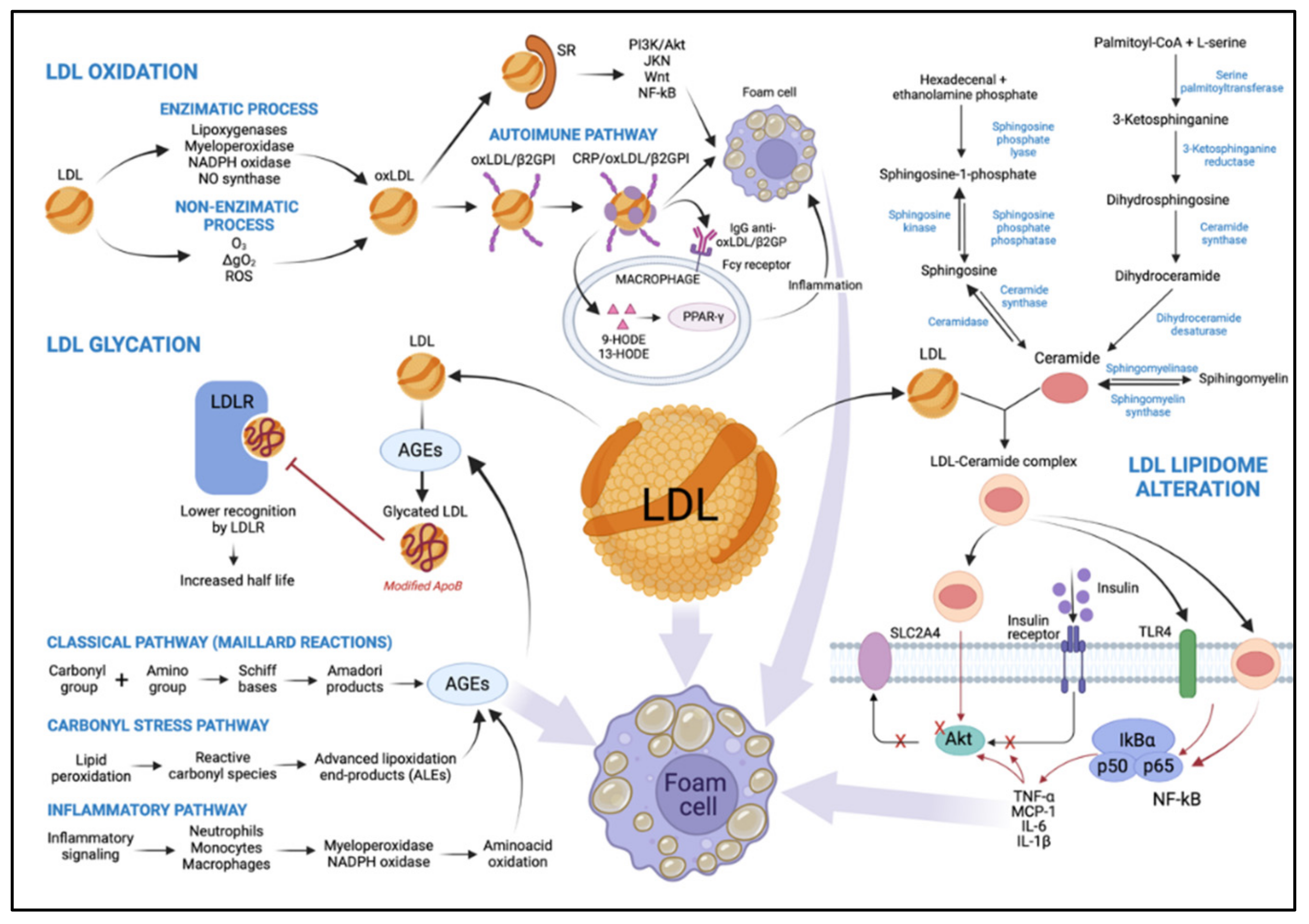
4.1. LDL Oxidation
4.1.1. LDL Oxidation by an Enzymatic Process
4.1.2. LDL Oxidation by Non-Enzymatic Process
4.2. Glycated LDL
4.3. Alteration of LDL Lipidome in T2DM: Ceramides
4.4. Deleterious Effects of LDL from T2DM Patients
4.5. Deleterious Effects of Modified LDL from T2DM Patients
4.6. Endothelial Dysfunction in Diabetes by Modified LDL
5. Potential Therapeutic Targets
5.1. Statins
5.2. Ezetimibe
5.3. Anti-PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Antibody (ab)
5.4. Increase LDL-C with SGLT2 Inhibitors
5.5. Insulin Treatment
5.6. Thiazolidinediones and sdLDL in T2DM
5.7. Glucagon-like Peptide-1 (GLP-1) Receptor Agonists, Liraglutide
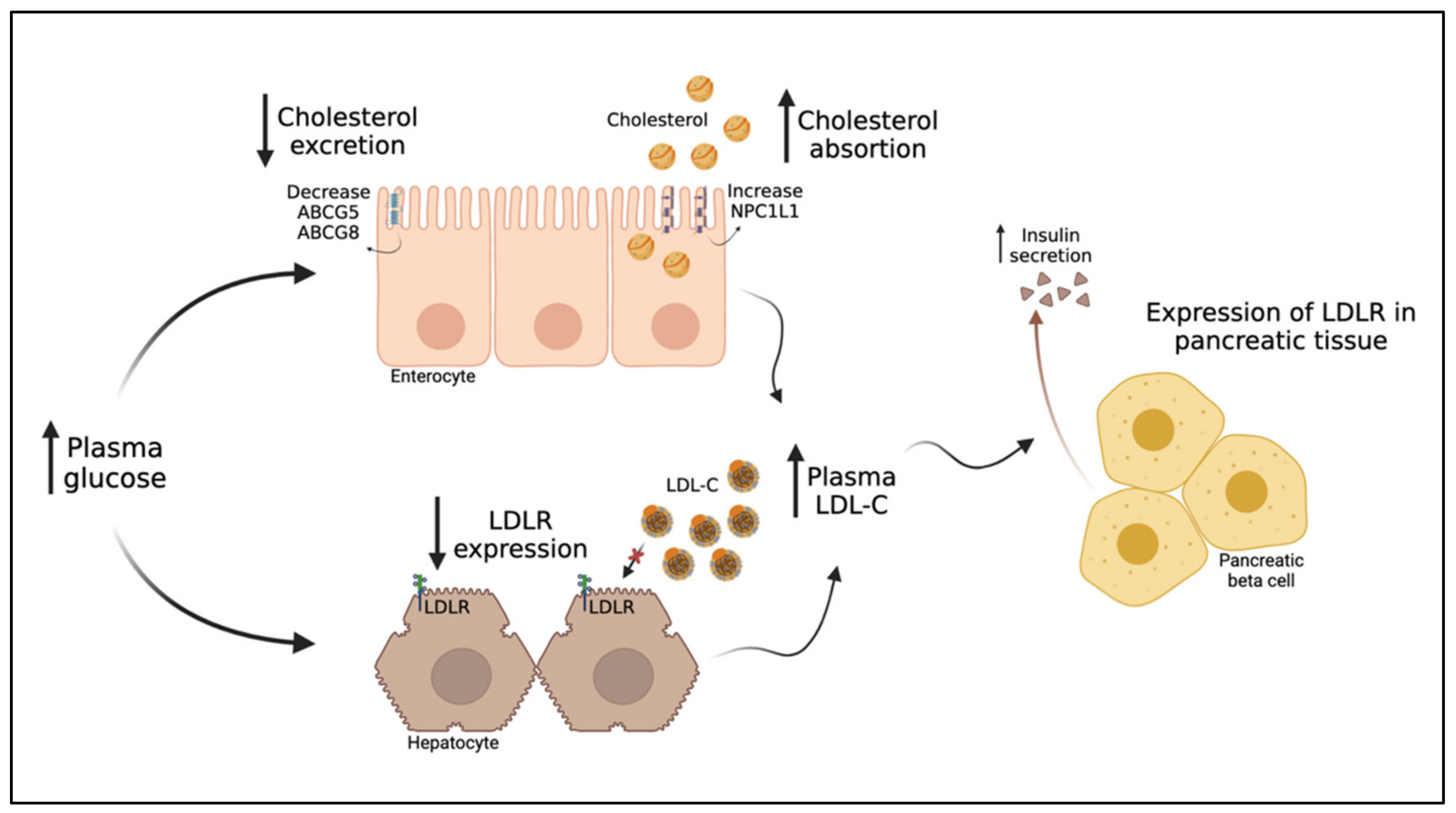
6. The Intriguing Inverse Relationship between LDL-C and T2DM
6.1. The Inverse Relationship between LDL-C and T2DM Risk
6.2. PCSK9 and HMGCR Variants Associated with LDL-C Reduction an Increased Risk of Diabetes
6.3. Reduced Risk of Diabetes in Familial Hypercholesterolemia
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| T2DM | Type 2 diabetes mellitus |
| CVD | Cardiovascular disease |
| apoB-100 | Apolipoprotein B-100 |
| HDL | High-density lipoprotein |
| LDL | Low-density lipoprotein |
| MM-LDL | Minimally oxidized LDL |
| Ox-LDL | Oxidized LDL |
| VLDL | Very-low-density lipoprotein |
| LPL | Lipoprotein lipase |
| CETP | Cholesteryl ester transfer protein |
| TG | Triglycerides |
| CE | Cholesteryl esters |
| FC | Free cholesterol |
| sdLDL | Small and dense LDL |
| LDLR | LDL receptors |
| IDL | Intermediate-density lipoprotein |
| apoA-I | Apolipoprotein A-I |
| HDL-C | HDL-cholesterol |
| DM | Diabetes mellitus |
| IMT | Intima-media thickness layer |
| LDL-C | LDL-cholesterol |
| NEFA | Non-esterified fatty acids |
| PI-3K | Phosphatidylinositol-3 kinase |
| MAPK | Mitogen-activated protein kinase |
| MTP | Microsomal TG-transfer protein |
| SRs | Scavenger receptors |
| SR-A1 | Class A1 scavenger receptor |
| LOX-1 | Lectin-like oxidized LDL receptor-1 |
| SREC | Scavenger receptor expressed by endothelial cell-I |
| SR-PSOX | Scavenger receptor for phosphatidylserine and oxidized lipoprotein |
| AKT | Protein kinase B |
| JNK | Janus kinase |
| NF-κB | Factor nuclear kappa B |
| β2ΓΠΙ | Beta2-glycoprotein I |
| CRP | C-reactive protein |
| TNF-α | Tumor necrosis factor-α |
| PDGF | Platelet-derived growth factor |
| PPAR-γ | Peroxisome proliferator-activated receptor γ |
| 9-HODE | 9-hydroxyoctadecadienoic acid |
| 13-HODE | 13-hydroxyoctadecadienoic acid |
| PUFA | Polyunsaturated fatty acids |
| HbAIC | Glycated hemoglobin |
| MPO | Myeloperoxidase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| HOCl | Hypochlorous acid |
| AGEs | Advanced glycation end products |
| O2− | Superoxide anion |
| ROS | Reactive oxygen species |
| NO | Nitric oxide |
| eNOS | Endothelial NO synthase |
| Lp-PLA2 | Lipoprotein-associated phospholipase A2 |
| 1ΔgO2 | Singlet oxygen |
| O3 | Ozone |
| PKC | Protein kinase C |
| ALEs | Advanced lipoxidation end-products |
| CER | Ceramides |
| IRS | Insulin receptor substrates |
| MMP | Metalloproteinases |
| THP-1 | Monocytic cell line |
| CREM | cAMP-responsive element modulator |
| ICER | Inducible cAMP early repressor |
| ER | Endoplasmic reticulum |
| PEDF | Pigment epithelium-derived factor |
| iNOS | Inducible NO synthase |
| MACEs | Major adverse cardiovascular events |
| NPC1L1 | Niemann–Pick C1-like 1 protein |
| SGLT2i | Sodium-glucose cotransporter 2 inhibitor |
| GLP-1 | Glucagon-like peptide- |
| DPP-4 | Dipeptidyl peptidase-4 |
| SLC2A4 | Insulin-sensitive solute carrier family 2, member 4 |
| HMGCR | 3-hydroxy-3-methylglutaryl-coenzyme A reductase |
| FH | Familial hypercholesterolemia |
References
- Turner, R.C.; Millns, H.; Neil, H.A.; Stratton, I.M.; Manley, S.E.; Matthews, D.R.; Holman, R.R. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998, 316, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Dannecker, C.; Wagner, R.; Peter, A.; Hummel, J.; Vosseler, A.; Häring, H.U.; Fritsche, A.; Birkenfeld, A.L.; Stefan, N.; Heni, M. Low-Density Lipoprotein Cholesterol Is Associated with Insulin Secretion. J. Clin. Endocrinol. Metab. 2021, 106, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Kopprasch, S.; Pietzsch, J.; Kuhlisch, E.; Fuecker, K.; Temelkova-Kurktschiev, T.; Hanefeld, M.; Kühne, H.; Julius, U.; Graessler, J. In vivo evidence for increased oxidation of circulating LDL in impaired glucose tolerance. Diabetes 2002, 51, 3102–3106. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004, 27, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Quehenberger, O.; Kondratenko, N.; Green, S.; Steinberg, D. Minimally oxidized low-density lipoprotein increases expression of scavenger receptor A, CD36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 794–802. [Google Scholar] [CrossRef]
- Njajou, O.T.; Kanaya, A.M.; Holvoet, P.; Connelly, S.; Strotmeyer, E.S.; Harris, T.B.; Cummings, S.R.; Hsueh, W.C.; Study, H.A. Association between oxidized LDL, obesity and type 2 diabetes in a population-based cohort, the Health, Aging and Body Composition Study. Diabetes Metab. Res. Rev. 2009, 25, 733–739. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M.; Bang, H.; Heiss, G.; Duncan, B.B.; Folsom, A.R.; Pankow, J.S. Circulating oxidised low-density lipoprotein and intercellular adhesion molecule-1 and risk of type 2 diabetes mellitus: The Atherosclerosis Risk in Communities Study. Diabetologia 2007, 50, 36–42. [Google Scholar] [CrossRef][Green Version]
- Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 816–823. [Google Scholar] [CrossRef]
- Basa, A.L.; Garber, A.J. Cardiovascular disease and diabetes: Modifying risk factors other than glucose control. Ochsner. J. 2001, 3, 132–137. [Google Scholar]
- Carmena, R.; Duriez, P.; Fruchart, J.C. Atherogenic lipoprotein particles in atherosclerosis. Circulation 2004, 109, 2–7. [Google Scholar] [CrossRef]
- Islam, S.U.; Ahmed, M.B.; Ahsan, H.; Lee, Y.S. Recent Molecular Mechanisms and Beneficial Effects of Phytochemicals and Plant-Based Whole Foods in Reducing LDL-C and Preventing Cardiovascular Disease. Antioxidants 2021, 10, 784. [Google Scholar] [CrossRef]
- Wadhera, R.K.; Steen, D.L.; Khan, I.; Giugliano, R.P.; Foody, J.M. A review of low-density lipoprotein cholesterol, treatment strategies, and its impact on cardiovascular disease morbidity and mortality. J. Clin. Lipidol. 2016, 10, 472–489. [Google Scholar] [CrossRef]
- Kobayashi, J.; Miyashita, K.; Nakajima, K.; Mabuchi, H. Hepatic Lipase: A Comprehensive View of Its Role on Plasma Lipid and Lipoprotein Metabolism. J. Atheroscler. Thromb. 2015, 22, 1001–1011. [Google Scholar] [CrossRef]
- Trinick, T.R.; Duly, E.B. Hyperlipidemia: Overview, 3rd ed.; Caballero, B., Ed.; Academic Press: Waltham, MA, USA, 2013; pp. 442–452. [Google Scholar]
- Walford, G.A. Lipoprotein Metabolism and the Treatment of Lipid Disorders. In Endocrinology: Adult and Pediatric, 7th ed.; Larry Jameson, J., de Groot, L.J., de Kretser, D.M., Giudice, L.C., Grossman, A.B., Melmed, S., Potts, J.T., Gordon, C.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 715–736. [Google Scholar]
- Hevonoja, T.; Pentikäinen, M.O.; Hyvönen, M.T.; Kovanen, P.T.; Ala-Korpela, M. Structure of low density lipoprotein (LDL) particles: Basis for understanding molecular changes in modified LDL. Biochim. Biophys. Acta 2000, 1488, 189–210. [Google Scholar] [CrossRef]
- Lin, J. Low-Density Lipoprotein: Biochemical and Metabolic Characteristics and Its Pathogenic Mechanism. In Apolipoproteins, Triglycerides and Cholesterol; Waisundara, V.Y., Jovandaric, M.Z., Eds.; IntechOpen: London, UK, 2020. [Google Scholar]
- Venugopal, S.K.; Anouro, M.; Jialal, I. Biochemistry, Low Density Lipoprotein. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Guerin, M.; Lassel, T.S.; Le Goff, W.; Farnier, M.; Chapman, M.J. Action of atorvastatin in combined hyperlipidemia: Preferential reduction of cholesteryl ester transfer from HDL to VLDL1 particles. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 189–197. [Google Scholar] [CrossRef]
- Chapman, M.J.; Goldstein, S.; Lagrange, D.; Laplaud, P.M. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J. Lipid. Res. 1981, 22, 339–358. [Google Scholar] [CrossRef]
- Dutheil, F.; Walther, G.; Chapier, R.; Mnatzaganian, G.; Lesourd, B.; Naughton, G.; Verney, J.; Fogli, A.; Sapin, V.; Duclos, M.; et al. Atherogenic subfractions of lipoproteins in the treatment of metabolic syndrome by physical activity and diet—The RESOLVE trial. Lipids Health Dis. 2014, 13, 112. [Google Scholar] [CrossRef]
- Garvey, W.T.; Kwon, S.; Zheng, D.; Shaughnessy, S.; Wallace, P.; Hutto, A.; Pugh, K.; Jenkins, A.J.; Klein, R.L.; Liao, Y. Effects of insulin resistance and type 2 diabetes on lipoprotein subclass particle size and concentration determined by nuclear magnetic resonance. Diabetes 2003, 52, 453–462. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Vergès, B. Lipid modification in type 2 diabetes: The role of LDL and HDL. Fundam. Clin. Pharmacol. 2009, 23, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.B. Transfer of low density lipoprotein into the arterial wall and risk of atherosclerosis. Atherosclerosis 1996, 123, 1–15. [Google Scholar] [CrossRef]
- Austin, M.A.; King, M.C.; Vranizan, K.M.; Krauss, R.M. Atherogenic lipoprotein phenotype. A proposed genetic marker for coronary heart disease risk. Circulation 1990, 82, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Austin, M.A.; Breslow, J.L.; Hennekens, C.H.; Buring, J.E.; Willett, W.C.; Krauss, R.M. Low-density lipoprotein subclass patterns and risk of myocardial infarction. JAMA 1988, 260, 1917–1921. [Google Scholar] [CrossRef]
- Galeano, N.F.; Milne, R.; Marcel, Y.L.; Walsh, M.T.; Levy, E.; Ngu’yen, T.D.; Gleeson, A.; Arad, Y.; Witte, L.; Al-Haideri, M.; et al. Apoprotein B structure and receptor recognition of triglyceride-rich low density lipoprotein (LDL) is modified in small LDL but not in triglyceride-rich LDL of normal size. J. Biol. Chem. 1994, 269, 511–519. [Google Scholar] [CrossRef]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef]
- Jansen, H.; Hop, W.; van Tol, A.; Bruschke, A.V.; Birkenhäger, J.C. Hepatic lipase and lipoprotein lipase are not major determinants of the low density lipoprotein subclass pattern in human subjects with coronary heart disease. Atherosclerosis 1994, 107, 45–54. [Google Scholar] [CrossRef]
- Campos, H.; Dreon, D.M.; Krauss, R.M. Associations of hepatic and lipoprotein lipase activities with changes in dietary composition and low density lipoprotein subclasses. J. Lipid Res. 1995, 36, 462–472. [Google Scholar] [CrossRef]
- Taskinen, M.R. Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia 2003, 46, 733–749. [Google Scholar] [CrossRef]
- Packard, C.J.; Shepherd, J. Lipoprotein heterogeneity and apolipoprotein B metabolism. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3542–3556. [Google Scholar] [CrossRef]
- Jin, J.L.; Zhang, H.W.; Cao, Y.X.; Liu, H.H.; Hua, Q.; Li, Y.F.; Zhang, Y.; Wu, N.Q.; Zhu, C.G.; Xu, R.X.; et al. Association of small dense low-density lipoprotein with cardiovascular outcome in patients with coronary artery disease and diabetes: A prospective, observational cohort study. Cardiovasc Diabetol 2020, 19, 45. [Google Scholar] [CrossRef]
- Inukai, T.; Yamamoto, R.; Suetsugu, M.; Matsumoto, S.; Wakabayashi, S.; Inukai, Y.; Matsutomo, R.; Takebayashi, K.; Aso, Y. Small low-density lipoprotein and small low-density lipoprotein/total low-density lipoprotein are closely associated with intima-media thickness of the carotid artery in Type 2 diabetic patients. J. Diabetes Complicat. 2005, 19, 269–275. [Google Scholar] [CrossRef]
- Gerber, P.A.; Thalhammer, C.; Schmied, C.; Spring, S.; Amann-Vesti, B.; Spinas, G.A.; Berneis, K. Small, dense LDL particles predict changes in intima media thickness and insulin resistance in men with type 2 diabetes and prediabetes—A prospective cohort study. PLoS ONE 2013, 8, e72763. [Google Scholar] [CrossRef]
- Krüger, M.; Babicz, K.; von Frieling-Salewsky, M.; Linke, W.A. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J. Mol. Cell Cardiol. 2010, 48, 910–916. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef]
- Schofield, J.D.; Liu, Y.; Rao-Balakrishna, P.; Malik, R.A.; Soran, H. Diabetes Dyslipidemia. Diabetes Ther. 2016, 7, 203–219. [Google Scholar] [CrossRef]
- Mooradian, A.D. Dyslipidemia in type 2 diabetes mellitus. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 150–159. [Google Scholar] [CrossRef]
- Guérin, M.; Le Goff, W.; Lassel, T.S.; Van Tol, A.; Steiner, G.; Chapman, M.J. Atherogenic role of elevated CE transfer from HDL to VLDL(1) and dense LDL in type 2 diabetes: Impact of the degree of triglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 282–288. [Google Scholar] [CrossRef]
- Jayaraman, S.; Pérez, A.; Miñambres, I.; Sánchez-Quesada, J.L.; Gursky, O. Heparin binding triggers human VLDL remodeling by circulating lipoprotein lipase: Relevance to VLDL functionality in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1867, 159064. [Google Scholar] [CrossRef]
- Sánchez-Quesada, J.L.; Pérez, A. Modified lipoproteins as biomarkers of cardiovascular risk in diabetes mellitus. Endocrinol. Nutr. 2013, 60, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Puig, N.; Montolio, L.; Camps-Renom, P.; Navarra, L.; Jiménez-Altayó, F.; Jiménez-Xarrié, E.; Sánchez-Quesada, J.L.; Benitez, S. Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages. Cells 2020, 9, 583. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quesada, J.L.; Benítez, S.; Ordóñez-Llanos, J. Electronegative low-density lipoprotein. Curr. Opin. Lipidol. 2004, 15, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kaneshi, T.; Ohta, T.; Saku, K. Relation between insulin resistance and fast-migrating LDL subfraction as characterized by capillary isotachophoresis. J. Lipid Res. 2005, 46, 2265–2277. [Google Scholar] [CrossRef]
- Nour Eldin, E.E.; Almarzouki, A.; Assiri, A.M.; Elsheikh, O.M.; Mohamed, B.E.; Babakr, A.T. Oxidized low density lipoprotein and total antioxidant capacity in type-2 diabetic and impaired glucose tolerance Saudi men. Diabetol. Metab. Syndr. 2014, 6, 94. [Google Scholar] [CrossRef]
- Steinberg, D.; Witztum, J.L. Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation 2002, 105, 2107–2111. [Google Scholar] [CrossRef]
- Harmon, M.E.; Campen, M.J.; Miller, C.; Shuey, C.; Cajero, M.; Lucas, S.; Pacheco, B.; Erdei, E.; Ramone, S.; Nez, T.; et al. Associations of Circulating Oxidized LDL and Conventional Biomarkers of Cardiovascular Disease in a Cross-Sectional Study of the Navajo Population. PLoS ONE 2016, 11, e0143102. [Google Scholar] [CrossRef]
- Alouffi, S.; Faisal, M.; Alatar, A.A.; Ahmad, S. Oxidative Modification of LDL by Various Physicochemical Techniques: Its Probable Role in Diabetes Coupled with CVDs. Biomed. Res. Int. 2018, 2018, 7390612. [Google Scholar] [CrossRef]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Volobueva, A.; Zhang, D.; Grechko, A.; Orekhov, A. Foam cell formation and cholesterol trafficking and metabolism disturbances in atherosclerosis. Cor Vasa 2018, 61, 48–55. [Google Scholar] [CrossRef]
- Tabuchi, M.; Inoue, K.; Usui-Kataoka, H.; Kobayashi, K.; Teramoto, M.; Takasugi, K.; Shikata, K.; Yamamura, M.; Ando, K.; Nishida, K.; et al. The association of C-reactive protein with an oxidative metabolite of LDL and its implication in atherosclerosis. J. Lipid Res. 2007, 48, 768–781. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M. Inflammation and atherosclerosis: Role of C-reactive protein in risk assessment. Am. J. Med. 2004, 116, 9–16. [Google Scholar] [CrossRef]
- Sui, X.; Liu, Y.; Li, Q.; Liu, G.; Song, X.; Su, Z.; Chang, X.; Zhou, Y.; Liang, B.; Huang, D. Oxidized low-density lipoprotein suppresses expression of prostaglandin E receptor subtype EP3 in human THP-1 macrophages. PLoS ONE 2014, 9, e110828. [Google Scholar] [CrossRef]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Moore, K.J.; Rosen, E.D.; Fitzgerald, M.L.; Randow, F.; Andersson, L.P.; Altshuler, D.; Milstone, D.S.; Mortensen, R.M.; Spiegelman, B.M.; Freeman, M.W. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 2001, 7, 41–47. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Colas, R.; Pruneta-Deloche, V.; Guichardant, M.; Luquain-Costaz, C.; Cugnet-Anceau, C.; Moret, M.; Vidal, H.; Moulin, P.; Lagarde, M.; Calzada, C. Increased lipid peroxidation in LDL from type-2 diabetic patients. Lipids 2010, 45, 723–731. [Google Scholar] [CrossRef]
- Ishino, S.; Mukai, T.; Kume, N.; Asano, D.; Ogawa, M.; Kuge, Y.; Minami, M.; Kita, T.; Shiomi, M.; Saji, H. Lectin-like oxidized LDL receptor-1 (LOX-1) expression is associated with atherosclerotic plaque instability—Analysis in hypercholesterolemic rabbits. Atherosclerosis 2007, 195, 48–56. [Google Scholar] [CrossRef]
- Li, L.; Renier, G. The oral anti-diabetic agent, gliclazide, inhibits oxidized LDL-mediated LOX-1 expression, metalloproteinase-9 secretion and apoptosis in human aortic endothelial cells. Atherosclerosis 2009, 204, 40–46. [Google Scholar] [CrossRef]
- Li, L.; Sawamura, T.; Renier, G. Glucose enhances endothelial LOX-1 expression: Role for LOX-1 in glucose-induced human monocyte adhesion to endothelium. Diabetes 2003, 52, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.K.; McNally, A.K.; Chisolm, G.M. Lipoxygenase-mediated transformation of human low density lipoprotein to an oxidized and cytotoxic complex. J. Lipid Res. 1991, 32, 63–70. [Google Scholar] [CrossRef]
- Takahashi, Y.; Zhu, H.; Yoshimoto, T. Essential roles of lipoxygenases in LDL oxidation and development of atherosclerosis. Antioxid. Redox. Signal 2005, 7, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Rankin, S.M.; Parthasarathy, S.; Steinberg, D. Evidence for a dominant role of lipoxygenase(s) in the oxidation of LDL by mouse peritoneal macrophages. J. Lipid Res. 1991, 32, 449–456. [Google Scholar] [CrossRef]
- Wen, Y.; Gu, J.; Chakrabarti, S.K.; Aylor, K.; Marshall, J.; Takahashi, Y.; Yoshimoto, T.; Nadler, J.L. The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-alpha in macrophages. Endocrinology 2007, 148, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Kühn, H.; Belkner, J.; Suzuki, H.; Yamamoto, S. Oxidative modification of human lipoproteins by lipoxygenases of different positional specificities. J. Lipid Res. 1994, 35, 1749–1759. [Google Scholar] [CrossRef]
- Funk, C.D.; Cyrus, T. 12/15-lipoxygenase, oxidative modification of LDL and atherogenesis. Trends Cardiovasc. Med. 2001, 11, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: Reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef]
- Pietzsch, J.; Lattke, P.; Julius, U. Oxidation of apolipoprotein B-100 in circulating LDL is related to LDL residence time. In vivo insights from stable-isotope studies. Arterioscler. Thromb. Vasc. Biol. 2000, 20, e63–e67. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M.; Etzioni, A.; Levy, R. Activation of NADPH oxidase required for macrophage-mediated oxidation of low-density lipoprotein. Metabolism 1996, 45, 1069–1079. [Google Scholar] [CrossRef]
- Keaney, J.F. Oxidative stress and the vascular wall: NADPH oxidases take center stage. Circulation 2005, 112, 2585–2588. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef]
- Gradinaru, D.; Borsa, C.; Ionescu, C.; Prada, G.I. Oxidized LDL and NO synthesis—Biomarkers of endothelial dysfunction and ageing. Mech. Ageing Dev. 2015, 151, 101–113. [Google Scholar] [CrossRef]
- Wang, W.; Hein, T.W.; Zhang, C.; Zawieja, D.C.; Liao, J.C.; Kuo, L. Oxidized low-density lipoprotein inhibits nitric oxide-mediated coronary arteriolar dilation by up-regulating endothelial arginase I. Microcirculation 2011, 18, 36–45. [Google Scholar] [CrossRef]
- Tellis, C.C.; Tselepis, A.D. The role of lipoprotein-associated phospholipase A2 in atherosclerosis may depend on its lipoprotein carrier in plasma. Biochim. Biophys. Acta 2009, 1791, 327–338. [Google Scholar] [CrossRef]
- Sánchez-Quesada, J.L.; Vinagre, I.; de Juan-Franco, E.; Sánchez-Hernández, J.; Blanco-Vaca, F.; Ordóñez-Llanos, J.; Pérez, A. Effect of improving glycemic control in patients with type 2 diabetes mellitus on low-density lipoprotein size, electronegative low-density lipoprotein and lipoprotein-associated phospholipase A2 distribution. Am. J. Cardiol. 2012, 110, 67–71. [Google Scholar] [CrossRef]
- Iuliano, L. Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 2011, 164, 457–468. [Google Scholar] [CrossRef]
- Heinecke, J.W.; Baker, L.; Rosen, H.; Chait, A. Superoxide-mediated modification of low density lipoprotein by arterial smooth muscle cells. J. Clin. Investig. 1986, 77, 757–761. [Google Scholar] [CrossRef]
- Fleming, I.; Mohamed, A.; Galle, J.; Turchanowa, L.; Brandes, R.P.; Fisslthaler, B.; Busse, R. Oxidized low-density lipoprotein increases superoxide production by endothelial nitric oxide synthase by inhibiting PKCalpha. Cardiovasc. Res. 2005, 65, 897–906. [Google Scholar] [CrossRef]
- Toma, L.; Stancu, C.S.; Sima, A.V. Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies. Biomedicines 2020, 9, 18. [Google Scholar] [CrossRef]
- Mol, M.; Degani, G.; Coppa, C.; Baron, G.; Popolo, L.; Carini, M.; Aldini, G.; Vistoli, G.; Altomare, A. Advanced lipoxidation end products (ALEs) as RAGE binders: Mass spectrometric and computational studies to explain the reasons why. Redox. Biol. 2019, 23, 101083. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.; Sharma, R.; Soran, H.; Charlton-Menys, V.; Elseweidy, M.; Durrington, P.N. Glycation as an atherogenic modification of LDL. Curr. Opin. Lipidol. 2008, 19, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Tames, F.J.; Mackness, M.I.; Arrol, S.; Laing, I.; Durrington, P.N. Non-enzymatic glycation of apolipoprotein B in the sera of diabetic and non-diabetic subjects. Atherosclerosis 1992, 93, 237–244. [Google Scholar] [CrossRef]
- Cohen, M.P.; Lautenslager, G.; Shea, E. Glycated LDL concentrations in non-diabetic and diabetic subjects measured with monoclonal antibodies reactive with glycated apolipoprotein B epitopes. Eur. J. Clin. Chem. Clin. Biochem. 1993, 31, 707–713. [Google Scholar] [CrossRef][Green Version]
- Kennedy, A.L.; Lyons, T.J. Glycation, oxidation, and lipoxidation in the development of diabetic complications. Metabolism 1997, 46, 14–21. [Google Scholar] [CrossRef]
- Sánchez-Quesada, J.L.; Vinagre, I.; De Juan-Franco, E.; Sánchez-Hernández, J.; Bonet-Marques, R.; Blanco-Vaca, F.; Ordóñez-Llanos, J.; Pérez, A. Impact of the LDL subfraction phenotype on Lp-PLA2 distribution, LDL modification and HDL composition in type 2 diabetes. Cardiovasc. Diabetol. 2013, 12, 112. [Google Scholar] [CrossRef]
- Rabbani, N.; Chittari, M.V.; Bodmer, C.W.; Zehnder, D.; Ceriello, A.; Thornalley, P.J. Increased glycation and oxidative damage to apolipoprotein B100 of LDL cholesterol in patients with type 2 diabetes and effect of metformin. Diabetes 2010, 59, 1038–1045. [Google Scholar] [CrossRef][Green Version]
- Hage Hassan, R.; Bourron, O.; Hajduch, E. Defect of insulin signal in peripheral tissues: Important role of ceramide. World J. Diabetes 2014, 5, 244–257. [Google Scholar] [CrossRef]
- Bandet, C.L.; Tan-Chen, S.; Bourron, O.; Le Stunff, H.; Hajduch, E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int. J. Mol. Sci. 2019, 20, 479. [Google Scholar] [CrossRef]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef]
- Wiesner, P.; Leidl, K.; Boettcher, A.; Schmitz, G.; Liebisch, G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J. Lipid Res. 2009, 50, 574–585. [Google Scholar] [CrossRef]
- Neeland, I.J.; Singh, S.; McGuire, D.K.; Vega, G.L.; Roddy, T.; Reilly, D.F.; Castro-Perez, J.; Kozlitina, J.; Scherer, P.E. Relation of plasma ceramides to visceral adiposity, insulin resistance and the development of type 2 diabetes mellitus: The Dallas Heart Study. Diabetologia 2018, 61, 2570–2579. [Google Scholar] [CrossRef]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.F.; Ferré, P.; et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 2018, 61, 399–412. [Google Scholar] [CrossRef]
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A.; et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep. 2017, 18, 2269–2279. [Google Scholar] [CrossRef]
- Boon, J.; Hoy, A.J.; Stark, R.; Brown, R.D.; Meex, R.C.; Henstridge, D.C.; Schenk, S.; Meikle, P.J.; Horowitz, J.F.; Kingwell, B.A.; et al. Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes 2013, 62, 401–410. [Google Scholar] [CrossRef]
- Nguyen, A.; Tao, H.; Metrione, M.; Hajri, T. Very low density lipoprotein receptor (VLDLR) expression is a determinant factor in adipose tissue inflammation and adipocyte-macrophage interaction. J. Biol. Chem. 2014, 289, 1688–1703. [Google Scholar] [CrossRef]
- Worley, J.R.; Hughes, D.A.; Dozio, N.; Gavrilovic, J.; Sampson, M.J. Low density lipoprotein from patients with Type 2 diabetes increases expression of monocyte matrix metalloproteinase and ADAM metalloproteinase genes. Cardiovasc. Diabetol. 2007, 6, 21. [Google Scholar] [CrossRef]
- Calzada, C.; Coulon, L.; Halimi, D.; Le Coquil, E.; Pruneta-Deloche, V.; Moulin, P.; Ponsin, G.; Véricel, E.; Lagarde, M. In vitro glycoxidized low-density lipoproteins and low-density lipoproteins isolated from type 2 diabetic patients activate platelets via p38 mitogen-activated protein kinase. J. Clin. Endocrinol. Metab. 2007, 92, 1961–1964. [Google Scholar] [CrossRef]
- Molina, C.A.; Foulkes, N.S.; Lalli, E.; Sassone-Corsi, P. Inducibility and negative autoregulation of CREM: An alternative promoter directs the expression of ICER, an early response repressor. Cell 1993, 75, 875–886. [Google Scholar] [CrossRef]
- Favre, D.; Niederhauser, G.; Fahmi, D.; Plaisance, V.; Brajkovic, S.; Beeler, N.; Allagnat, F.; Haefliger, J.A.; Regazzi, R.; Waeber, G.; et al. Role for inducible cAMP early repressor in promoting pancreatic beta cell dysfunction evoked by oxidative stress in human and rat islets. Diabetologia 2011, 54, 2337–2346. [Google Scholar] [CrossRef][Green Version]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef]
- Fu, D.; Wu, M.; Zhang, J.; Du, M.; Yang, S.; Hammad, S.M.; Wilson, K.; Chen, J.; Lyons, T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 2012, 55, 3128–3140. [Google Scholar] [CrossRef]
- Fu, D.; Yu, J.Y.; Wu, M.; Du, M.; Chen, Y.; Abdelsamie, S.A.; Li, Y.; Chen, J.; Boulton, M.E.; Ma, J.X.; et al. Immune complex formation in human diabetic retina enhances toxicity of oxidized LDL towards retinal capillary pericytes. J. Lipid Res. 2014, 55, 860–869. [Google Scholar] [CrossRef]
- Hadi, H.A.; Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 2007, 3, 853–876. [Google Scholar]
- Artwohl, M.; Graier, W.F.; Roden, M.; Bischof, M.; Freudenthaler, A.; Waldhäusl, W.; Baumgartner-Parzer, S.M. Diabetic LDL triggers apoptosis in vascular endothelial cells. Diabetes 2003, 52, 1240–1247. [Google Scholar] [CrossRef][Green Version]
- Schalkwijk, C.G.; Stehouwer, C.D. Vascular complications in diabetes mellitus: The role of endothelial dysfunction. Clin. Sci. 2005, 109, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Toma, L.; Stancu, C.S.; Sanda, G.M.; Sima, A.V. Anti-oxidant and anti-inflammatory mechanisms of amlodipine action to improve endothelial cell dysfunction induced by irreversibly glycated LDL. Biochem. Biophys. Res. Commun. 2011, 411, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.S.; Georgescu, A.; Toma, L.; Sanda, G.M.; Sima, A.V. Glycated low density lipoproteins alter vascular reactivity in hyperlipidemic hyperglycemic hamsters. Ann. Rom. Soc. Cell Biol. 2012, 17, 9–15. [Google Scholar]
- Evans, M.; Khan, N.; Rees, A. Diabetic dyslipidaemia and coronary heart disease: New perspectives. Curr. Opin. Lipidol. 1999, 10, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Posch, K.; Simecek, S.; Wascher, T.C.; Jürgens, G.; Baumgartner-Parzer, S.; Kostner, G.M.; Graier, W.F. Glycated low-density lipoprotein attenuates shear stress-induced nitric oxide synthesis by inhibition of shear stress-activated l-arginine uptake in endothelial cells. Diabetes 1999, 48, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction-a major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef]
- Rizzo, M.; Berneis, K.; Koulouris, S.; Pastromas, S.; Rini, G.B.; Sakellariou, D.; Manolis, A.S. Should we measure routinely oxidised and atherogenic dense low-density lipoproteins in subjects with type 2 diabetes? Int. J. Clin. Pract. 2010, 64, 1632–1642. [Google Scholar] [CrossRef]
- Miller, P.E.; Martin, S.S. Approach to Statin Use in 2016: An Update. Curr. Atheroscler. Rep. 2016, 18, 20. [Google Scholar] [CrossRef]
- Adiga, K.I.; Padmakumar, R.; Shashikiran, U. A survey on intensity of statin therapy among diabetes mellitus patients in secondary care practice. Int. J. Diabetes Dev. Ctries. 2020, 40, 607–611. [Google Scholar] [CrossRef]
- Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; Simes, J.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef]
- Kearney, P.M.; Blackwell, L.; Collins, R.; Keech, A.; Simes, J.; Peto, R.; Armitage, J.; Baigent, C. Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: A meta-analysis. Lancet 2008, 371, 117–125. [Google Scholar] [CrossRef]
- Elnaem, M.H.; Mohamed, M.H.N.; Huri, H.Z.; Azarisman, S.M.; Elkalmi, R.M. Statin Therapy Prescribing for Patients with Type 2 Diabetes Mellitus: A Review of Current Evidence and Challenges. J. Pharm. Bioallied. Sci. 2017, 9, 80–87. [Google Scholar] [CrossRef]
- Vergès, B.; Florentin, E.; Baillot-Rudoni, S.; Monier, S.; Petit, J.M.; Rageot, D.; Gambert, P.; Duvillard, L. Effects of 20 mg rosuvastatin on VLDL1-, VLDL2-, IDL- and LDL-ApoB kinetics in type 2 diabetes. Diabetologia 2008, 51, 1382–1390. [Google Scholar] [CrossRef]
- Collins, R.; Armitage, J.; Parish, S.; Sleigh, P.; Peto, R.; Group HPSC. MRC/BHF Heart Protection Study of Cholesterol-Lowering with Simvastatin in 5963 People with Diabetes: A Randomised Placebo-Controlled Trial. Lancet 2003, 361, 2005–2016. [Google Scholar] [CrossRef]
- Jialal, I.; Singh, G. Management of diabetic dyslipidemia: An update. World J. Diabetes 2019, 10, 280–290. [Google Scholar] [CrossRef]
- Takemoto, M.; Liao, J.K. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Patti, A.M.; Giglio, R.V.; Papanas, N.; Rizzo, M.; Rizvi, A.A. Future perspectives of the pharmacological management of diabetic dyslipidemia. Expert Rev. Clin. Pharmacol. 2019, 12, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Phan, B.A.; Dayspring, T.D.; Toth, P.P. Ezetimibe therapy: Mechanism of action and clinical update. Vasc. Health Risk Manag. 2012, 8, 415–427. [Google Scholar] [CrossRef]
- Leiter, L.A.; Betteridge, D.J.; Farnier, M.; Guyton, J.R.; Lin, J.; Shah, A.; Johnson-Levonas, A.O.; Brudi, P. Lipid-altering efficacy and safety profile of combination therapy with ezetimibe/statin vs. statin monotherapy in patients with and without diabetes: An analysis of pooled data from 27 clinical trials. Diabetes Obes. Metab. 2011, 13, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Ravid, Z.; Bendayan, M.; Delvin, E.; Sane, A.T.; Elchebly, M.; Lafond, J.; Lambert, M.; Mailhot, G.; Levy, E. Modulation of intestinal cholesterol absorption by high glucose levels: Impact on cholesterol transporters, regulatory enzymes, and transcription factors. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 295, G873–G885. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, P.; Boddy, C.S.; Soni, V.; Saksena, S.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. D-Glucose modulates intestinal Niemann-Pick C1-like 1 (NPC1L1) gene expression via transcriptional regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G203–G210. [Google Scholar] [CrossRef]
- Lally, S.; Tan, C.Y.; Owens, D.; Tomkin, G.H. Messenger RNA levels of genes involved in dysregulation of postprandial lipoproteins in type 2 diabetes: The role of Niemann-Pick C1-like 1, ATP-binding cassette, transporters G5 and G8, and of microsomal triglyceride transfer protein. Diabetologia 2006, 49, 1008–1016. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Cannon, C.P.; Blazing, M.A.; Nicolau, J.C.; Corbalán, R.; Špinar, J.; Park, J.G.; White, J.A.; Bohula, E.A.; Braunwald, E.; et al. Benefit of Adding Ezetimibe to Statin Therapy on Cardiovascular Outcomes and Safety in Patients With Versus Without Diabetes Mellitus: Results From IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial). Circulation 2018, 137, 1571–1582. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- Raedler, L.A. Praluent (Alirocumab): First PCSK9 Inhibitor Approved by the FDA for Hypercholesterolemia. Am. Health Drug Benefits 2016, 9, 123–126. [Google Scholar]
- Taskinen, M.R.; Björnson, E.; Kahri, J.; Söderlund, S.; Matikainen, N.; Porthan, K.; Ainola, M.; Hakkarainen, A.; Lundbom, N.; Fermanelli, V.; et al. Effects of Evolocumab on the Postprandial Kinetics of Apo (Apolipoprotein) B100-and B48-Containing Lipoproteins in Subjects with Type 2 Diabetes. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 962–975. [Google Scholar] [CrossRef]
- Zhang, P.; Gao, J.; Pu, C.; Zhang, Y. Apolipoprotein status in type 2 diabetes mellitus and its complications (Review). Mol. Med. Rep. 2017, 16, 9279–9286. [Google Scholar] [CrossRef]
- Basu, D.; Huggins, L.A.; Scerbo, D.; Obunike, J.; Mullick, A.E.; Rothenberg, P.L.; Di Prospero, N.A.; Eckel, R.H.; Goldberg, I.J. Mechanism of Increased LDL (Low-Density Lipoprotein) and Decreased Triglycerides With SGLT2 (Sodium-Glucose Cotransporter 2) Inhibition. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2207–2216. [Google Scholar] [CrossRef]
- Breder, I.; Cunha Breder, J.; Bonilha, I.; Munhoz, D.B.; Medorima, S.T.K.; Oliveira, D.C.; do Carmo, H.R.; Moreira, C.; Kontush, A.; Zimetti, F.; et al. Rationale and design of the expanded combination of evolocumab plus empagliflozin in diabetes: EXCEED-BHS3 trial. Ther. Adv. Chronic. Dis. 2020, 11, 2040622320959248. [Google Scholar] [CrossRef]
- Schuster, S.; Rubil, S.; Endres, M.; Princen, H.M.G.; Boeckel, J.N.; Winter, K.; Werner, C.; Laufs, U. Anti-PCSK9 antibodies inhibit pro-atherogenic mechanisms in APOE*3Leiden.CETP mice. Sci. Rep. 2019, 9, 11079. [Google Scholar] [CrossRef]
- Li, H.L.; Lip, G.H.; Feng, Q.; Fei, Y.; Tse, Y.K.; Wu, M.Z.; Ren, Q.W.; Tse, H.F.; Cheung, B.Y.; Yiu, K.H. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2021, 20, 100. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Zaccardi, F.; Webb, D.R.; Htike, Z.Z.; Youssef, D.; Khunti, K.; Davies, M.J. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: Systematic review and network meta-analysis. Diabetes Obes. Metab. 2016, 18, 783–794. [Google Scholar] [CrossRef]
- Bode, B.; Stenlöf, K.; Harris, S.; Sullivan, D.; Fung, A.; Usiskin, K.; Meininger, G. Long-term efficacy and safety of canagliflozin over 104 weeks in patients aged 55–80 years with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 294–303. [Google Scholar] [CrossRef]
- Inagaki, N.; Harashima, S.; Maruyama, N.; Kawaguchi, Y.; Goda, M.; Iijima, H. Efficacy and safety of canagliflozin in combination with insulin: A double-blind, randomized, placebo-controlled study in Japanese patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2016, 15, 89. [Google Scholar] [CrossRef]
- Matthaei, S.; Bowering, K.; Rohwedder, K.; Sugg, J.; Parikh, S.; Johnsson, E.; Group, S. Durability and tolerability of dapagliflozin over 52 weeks as add-on to metformin and sulphonylurea in type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B.; Association, A.D.; Diabetes, E.A.f.S.o. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy: A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef]
- Duvillard, L.; Pont, F.; Florentin, E.; Gambert, P.; Vergès, B. Significant improvement of apolipoprotein B-containing lipoprotein metabolism by insulin treatment in patients with non-insulin-dependent diabetes mellitus. Diabetologia 2000, 43, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Hirano, T.; Yamamoto, T.; Ito, Y.; Adachi, M. Intensive insulin therapy reduces small dense low-density lipoprotein particles in patients with type 2 diabetes mellitus: Relationship to triglyceride-rich lipoprotein subspecies. Metabolism 2006, 55, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Duvillard, L.; Florentin, E.; Lizard, G.; Petit, J.M.; Galland, F.; Monier, S.; Gambert, P.; Vergès, B. Cell surface expression of LDL receptor is decreased in type 2 diabetic patients and is normalized by insulin therapy. Diabetes Care 2003, 26, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Marx, N.; Duez, H.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors and atherogenesis: Regulators of gene expression in vascular cells. Circ. Res. 2004, 94, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.M.; Reid, J.; Taylor, G.J.; Stirling, C.; Reckless, J.P. Favorable effects of pioglitazone and metformin compared with gliclazide on lipoprotein subfractions in overweight patients with early type 2 diabetes. Diabetes Care 2004, 27, 41–46. [Google Scholar] [CrossRef]
- Perez, A.; Khan, M.; Johnson, T.; Karunaratne, M. Pioglitazone plus a sulphonylurea or metformin is associated with increased lipoprotein particle size in patients with type 2 diabetes. Diab. Vasc. Dis. Res. 2004, 1, 44–50. [Google Scholar] [CrossRef]
- Deeg, M.A.; Buse, J.B.; Goldberg, R.B.; Kendall, D.M.; Zagar, A.J.; Jacober, S.J.; Khan, M.A.; Perez, A.T.; Tan, M.H.; Investigators, G.S. Pioglitazone and rosiglitazone have different effects on serum lipoprotein particle concentrations and sizes in patients with type 2 diabetes and dyslipidemia. Diabetes Care 2007, 30, 2458–2464. [Google Scholar] [CrossRef]
- Kintscher, U. Pharmacological differences of glitazones: Does peroxisome proliferator-activated receptor-alpha activation make the difference? J. Am. Coll. Cardiol. 2008, 52, 882–884. [Google Scholar] [CrossRef][Green Version]
- Berneis, K.; Rizzo, M.; Stettler, C.; Chappuis, B.; Braun, M.; Diem, P.; Christ, E.R. Comparative effects of rosiglitazone and pioglitazone on fasting and postprandial low-density lipoprotein size and subclasses in patients with Type 2 diabetes. Expert Opin. Pharmacother. 2008, 9, 343–349. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- Garber, A.J. Long-acting glucagon-like peptide 1 receptor agonists: A review of their efficacy and tolerability. Diabetes Care 2011, 34, S279–S284. [Google Scholar] [CrossRef]
- Vergès, B.; Duvillard, L.; Pais de Barros, J.P.; Bouillet, B.; Baillot-Rudoni, S.; Rouland, A.; Petit, J.M.; Degrace, P.; Demizieux, L. Liraglutide Increases the Catabolism of Apolipoprotein B100-Containing Lipoproteins in Patients with Type 2 Diabetes and Reduces Proprotein Convertase Subtilisin/Kexin Type 9 Expression. Diabetes Care 2021, 44, 1027–1037. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Nikolic, D.; Giglio, R.V.; Rizvi, A.A.; Patti, A.M.; Montalto, G.; Maranta, F.; Cianflone, D.; Stoian, A.P.; Rizzo, M. Liraglutide Reduces Carotid Intima-Media Thickness by Reducing Small Dense Low-Density Lipoproteins in a Real-World Setting of Patients with Type 2 Diabetes: A Novel Anti-Atherogenic Effect. Diabetes Ther. 2021, 12, 261–274. [Google Scholar] [CrossRef]
- Rizzo, M.; Nikolic, D.; Patti, A.M.; Mannina, C.; Montalto, G.; McAdams, B.S.; Rizvi, A.A.; Cosentino, F. GLP-1 receptor agonists and reduction of cardiometabolic risk: Potential underlying mechanisms. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2814–2821. [Google Scholar] [CrossRef]
- Fall, T.; Xie, W.; Poon, W.; Yaghootkar, H.; Mägi, R.; Knowles, J.W.; Lyssenko, V.; Weedon, M.; Frayling, T.M.; Ingelsson, E.; et al. Using Genetic Variants to Assess the Relationship Between Circulating Lipids and Type 2 Diabetes. Diabetes 2015, 64, 2676–2684. [Google Scholar] [CrossRef]
- Andersson, C.; Lyass, A.; Larson, M.G.; Robins, S.J.; Vasan, R.S. Low-density-lipoprotein cholesterol concentrations and risk of incident diabetes: Epidemiological and genetic insights from the Framingham Heart Study. Diabetologia 2015, 58, 2774–2780. [Google Scholar] [CrossRef]
- Feng, Q.; Wei, W.Q.; Chung, C.P.; Levinson, R.T.; Sundermann, A.C.; Mosley, J.D.; Bastarache, L.; Ferguson, J.F.; Cox, N.J.; Roden, D.M.; et al. Relationship between very low low-density lipoprotein cholesterol concentrations not due to statin therapy and risk of type 2 diabetes: A US-based cross-sectional observational study using electronic health records. PLoS Med 2018, 15, e1002642. [Google Scholar] [CrossRef]
- Cai, R.; Yuan, Y.; Zhou, Y.; Xia, W.; Wang, P.; Sun, H.; Yang, Y.; Huang, R.; Wang, S. Lower intensified target LDL-c level of statin therapy results in a higher risk of incident diabetes: A meta-analysis. PLoS ONE 2014, 9, e104922. [Google Scholar] [CrossRef]
- Everett, B.M.; Mora, S.; Glynn, R.J.; MacFadyen, J.; Ridker, P.M. Safety profile of subjects treated to very low low-density lipoprotein cholesterol levels (<30 mg/dl) with rosuvastatin 20 mg daily (from JUPITER). Am. J. Cardiol. 2014, 114, 1682–1689. [Google Scholar] [CrossRef]
- Sattar, N.; Preiss, D.; Murray, H.M.; Welsh, P.; Buckley, B.M.; de Craen, A.J.; Seshasai, S.R.; McMurray, J.J.; Freeman, D.J.; Jukema, J.W.; et al. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Lancet 2010, 375, 735–742. [Google Scholar] [CrossRef]
- De Carvalho, L.S.F.; Campos, A.M.; Sposito, A.C. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibitors and Incident Type 2 Diabetes: A Systematic Review and Meta-analysis With Over 96,000 Patient-Years. Diabetes Care 2018, 41, 364–367. [Google Scholar] [CrossRef]
- Preiss, D.; Seshasai, S.R.; Welsh, P.; Murphy, S.A.; Ho, J.E.; Waters, D.D.; DeMicco, D.A.; Barter, P.; Cannon, C.P.; Sabatine, M.S.; et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: A meta-analysis. JAMA 2011, 305, 2556–2564. [Google Scholar] [CrossRef]
- Wang, S.; Cai, R.; Yuan, Y.; Varghese, Z.; Moorhead, J.; Ruan, X.Z. Association between reductions in low-density lipoprotein cholesterol with statin therapy and the risk of new-onset diabetes: A meta-analysis. Sci. Rep. 2017, 7, 39982. [Google Scholar] [CrossRef]
- Waters, D.D.; Ho, J.E.; Boekholdt, S.M.; DeMicco, D.A.; Kastelein, J.J.; Messig, M.; Breazna, A.; Pedersen, T.R. Cardiovascular event reduction versus new-onset diabetes during atorvastatin therapy: Effect of baseline risk factors for diabetes. J. Am. Coll. Cardiol. 2013, 61, 148–152. [Google Scholar] [CrossRef]
- Sattar, N.; Taskinen, M.R. Statins are diabetogenic—Myth or reality? Atheroscler. Suppl. 2012, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Besseling, J.; Kastelein, J.J.; Defesche, J.C.; Hutten, B.A.; Hovingh, G.K. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA 2015, 313, 1029–1036. [Google Scholar] [CrossRef]
- Sampson, U.K.; Linton, M.F.; Fazio, S. Are statins diabetogenic? Curr. Opin. Cardiol. 2011, 26, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Hannaert, J.C.; Grupping, A.Y.; Pipeleers, D.G. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 2002, 143, 3449–3453. [Google Scholar] [CrossRef] [PubMed]
- Nakata, M.; Nagasaka, S.; Kusaka, I.; Matsuoka, H.; Ishibashi, S.; Yada, T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): Implications in glycaemic control. Diabetologia 2006, 49, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Robinson, J.G.; Brook, R.D.; Catapano, A.L.; Chapman, M.J.; Neff, D.R.; Voros, S.; Giugliano, R.P.; Davey Smith, G.; Fazio, S.; et al. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N. Engl. J. Med. 2016, 375, 2144–2153. [Google Scholar] [CrossRef]
- Schmidt, A.F.; Swerdlow, D.I.; Holmes, M.V.; Patel, R.S.; Fairhurst-Hunter, Z.; Lyall, D.M.; Hartwig, F.P.; Horta, B.L.; Hyppönen, E.; Power, C.; et al. PCSK9 genetic variants and risk of type 2 diabetes: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2017, 5, 97–105. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Preiss, D.; Kuchenbaecker, K.B.; Holmes, M.V.; Engmann, J.E.; Shah, T.; Sofat, R.; Stender, S.; Johnson, P.C.; Scott, R.A.; et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: Evidence from genetic analysis and randomised trials. Lancet 2015, 385, 351–361. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Climent, E.; Pérez-Calahorra, S.; Marco-Benedí, V.; Plana, N.; Sánchez, R.; Ros, E.; Ascaso, J.F.; Puzo, J.; Almagro, F.; Lahoz, C.; et al. Effect of LDL cholesterol, statins and presence of mutations on the prevalence of type 2 diabetes in heterozygous familial hypercholesterolemia. Sci. Rep. 2017, 7, 5596. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonilha, I.; Hajduch, E.; Luchiari, B.; Nadruz, W.; Le Goff, W.; Sposito, A.C. The Reciprocal Relationship between LDL Metabolism and Type 2 Diabetes Mellitus. Metabolites 2021, 11, 807. https://doi.org/10.3390/metabo11120807
Bonilha I, Hajduch E, Luchiari B, Nadruz W, Le Goff W, Sposito AC. The Reciprocal Relationship between LDL Metabolism and Type 2 Diabetes Mellitus. Metabolites. 2021; 11(12):807. https://doi.org/10.3390/metabo11120807
Chicago/Turabian StyleBonilha, Isabella, Eric Hajduch, Beatriz Luchiari, Wilson Nadruz, Wilfried Le Goff, and Andrei C. Sposito. 2021. "The Reciprocal Relationship between LDL Metabolism and Type 2 Diabetes Mellitus" Metabolites 11, no. 12: 807. https://doi.org/10.3390/metabo11120807
APA StyleBonilha, I., Hajduch, E., Luchiari, B., Nadruz, W., Le Goff, W., & Sposito, A. C. (2021). The Reciprocal Relationship between LDL Metabolism and Type 2 Diabetes Mellitus. Metabolites, 11(12), 807. https://doi.org/10.3390/metabo11120807