
Nerpa: A Tool for Discovering Biosynthetic Gene Clusters of Bacterial Nonribosomal Peptides

 , , , , , ,
, , , , , ,  and
and 
Abstract
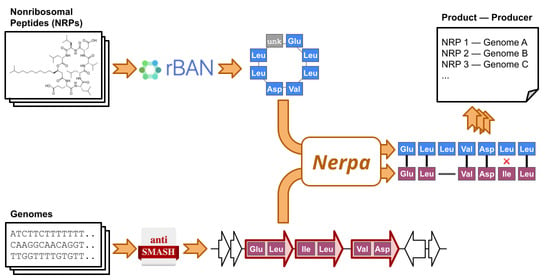
1. Introduction
2. Results
2.1. Outline of the Nerpa Pipeline
2.2. Database of Putative NRPs
2.3. Benchmarking Nerpa Accuracy
2.4. Benchmarking Nerpa Performance
2.5. Linking Known and Novel BGCs to Their Products with Nerpa
2.6. Experimental Validation of Putative Ngercheumicin BGC
3. Discussion
4. Materials and Methods
4.1. Processing of NRP Structures
4.1.1. Monomer Graph Construction
4.1.2. Monomer Post-Processing
4.1.3. Linear Representation of Monomer Graphs
4.2. Processing of Genome Sequences
4.2.1. BGC Monomers
4.2.2. Monomer Strips
4.2.3. BGC Splitting
4.2.4. Handling of NRPS Assembly Lines
4.3. Scoring of Monomer Sequences
4.3.1. General Notations
4.3.2. Nerpa Score Summary
4.3.3. Scoring Matches and Mismatches
4.3.4. Scoring Indels
4.3.5. Learning Nerpa Parameters
4.4. Reporting of Results
4.5. Software Implementation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BGC | biosynthetic gene cluster |
| FDR | false discovery rate |
| GB | gigabyte |
| GNPS | Global Natural Product Social molecular networking |
| MS/MS | tandem mass spectra |
| NCBI | National Center for Biotechnology Information |
| NRP | nonribosomal peptide |
| NRPS | nonribosomal peptide synthetase |
| RAM | random access memory (computer memory) |
| RefSeq | NCBI Reference Sequence Database |
| RiPP | ribosomally synthesized and post-translationally modified peptide |
| SD | standard deviation |
Appendix A. Running Commands
- PRISM
- version 2.1.5, downloaded from https://github.com/magarveylab/prism-releases/releases/download/v2.1.5/prism.jar (accessed on 30 September 2021)java -jar <PATH_TO_PRISM_JAR> -a -p \-f <PATH_TO_GENOME_FASTA> -w 10000 \-tt -o <OUTPUT_DIR> -r <PATH_TO_WEBCONTENT>
- GRAPE
- version 2.9.1, downloaded from https://github.com/magarveylab/grape-release/archive/refs/tags/2.9.1.tar.gz (accessed on 30 September 2021)java -jar <PATH_TO_GRAPE_JAR> -s <SMILES_STRING> \-img -json -txt -o <OUTPUT_DIR>
- GARLIC
- version 1.0.2, downloaded from https://github.com/magarveylab/garlic-release/archive/refs/tags/1.0.2.tar.gz (accessed on 30 September 2021)java -jar <PATH_TO_GARLIC_JAR> -q <PATH_TO_PRISM_OUT> \-a <PATH_TO_GRAPE_OUT> -o <OUTPUT_DIR>
- antiSMASH
- version 5.2.0, downloaded from https://dl.secondarymetabolites.org/releases/5.2.0/antismash-5.2.0.tar.gz (accessed on 30 September 2021)run_antismash.py <PATH_TO_GENOME_FASTA> \--genefinding-tool prodigal --minimal \--skip-zip-file --enable-nrps-pks \--output-dir <OUTPUT_DIR>
- rBAN
- version 1.0, downloaded from https://bitbucket.org/sib-pig/rban/downloads/rBAN-1.0.jar (accessed on 30 September 2021)java -jar <PATH_TO_RBAN_JAR> \-monomersDB <PATH_TO_MONOMERS> \-inputFile <PATH_TO_INPUT_JSON> \-outputFileName <OUTPUT_FILE_NAME> \-outputFolder <OUTPUT_DIR>
- Nerpa
- version 1.0.0, downloaded from https://github.com/ablab/nerpa/releases/tag/v1.0.0 (accessed on 30 September 2021)nerpa.py -a <PATH_TO_ANTISMASH_OUTPUTS> \--smiles-tsv <PATH_TO_STRUCTURES> \--col-smiles <COL_SMILES> --col-id <COL_ID> \--sep <SEP> --process-hybrids -o <OUTPUT_DIR>
Appendix B. MS/MS Experiment Details
Appendix B.1. Microorganism Culturing
Appendix B.2. Extraction of Metabolites
Appendix B.3. Sample Preparation and LC-MS/MS Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Collision RF | Transfer Time | Collision |
|---|---|---|---|
| 0 | 450.0 | 70.0 | 125 |
| 25 | 550.0 | 75.0 | 100 |
| 50 | 800.0 | 90.0 | 100 |
| 75 | 1100.0 | 95.0 | 75 |
| Type | Mass | Width | Collision | Charge State |
|---|---|---|---|---|
| Base | 100.00 | 4.00 | 22.00 | 1 |
| Base | 100.00 | 4.00 | 18.00 | 2 |
| Base | 300.00 | 5.00 | 27.00 | 1 |
| Base | 300.00 | 5.00 | 22.00 | 2 |
| Base | 500.00 | 6.00 | 35.00 | 1 |
| Base | 500.00 | 6.00 | 30.00 | 2 |
| Base | 1000.00 | 8.00 | 45.00 | 1 |
| Base | 1000.00 | 8.00 | 35.00 | 2 |
| Base | 2000.00 | 10.00 | 50.00 | 1 |
| Base | 2000.00 | 10.00 | 50.00 | 2 |
References
- Agrawal, S.; Acharya, D.; Adholeya, A.; Barrow, C.J.; Deshmukh, S.K. Nonribosomal peptides from marine microbes and their antimicrobial and anticancer potential. Front. Pharmacol. 2017, 8, 828. [Google Scholar] [CrossRef]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Bull. World Health Organ. 1929, 79, 780–790. [Google Scholar] [CrossRef]
- Flissi, A.; Dufresne, Y.; Michalik, J.; Tonon, L.; Janot, S.; Noé, L.; Jacques, P.; Leclère, V.; Pupin, M. Norine, the knowledgebase dedicated to non-ribosomal peptides, is now open to crowdsourcing. Nucleic Acids Res. 2016, 44, D1113–D1118. [Google Scholar] [CrossRef]
- Marahiel, M.A.; Stachelhaus, T.; Mootz, H.D. Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis. Chem. Rev. 1997, 97, 2651–2674. [Google Scholar] [CrossRef] [PubMed]
- Stachelhaus, T.; Mootz, H.D.; Marahiel, M.A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999, 6, 493–505. [Google Scholar] [CrossRef]
- Dejong, C.A.; Chen, G.M.; Li, H.; Johnston, C.W.; Edwards, M.R.; Rees, P.N.; Skinnider, M.A.; Webster, A.L.; Magarvey, N.A. Polyketide and nonribosomal peptide retro-biosynthesis and global gene cluster matching. Nat. Chem. Biol. 2016, 12, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, D.F. Cracking the nonribosomal code. Cell Chem. Biol. 2016, 23, 535–537. [Google Scholar] [CrossRef][Green Version]
- Rausch, C.; Hoof, I.; Weber, T.; Wohlleben, W.; Huson, D.H. Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol. Biol. 2007, 7, 1–15. [Google Scholar] [CrossRef]
- Li, M.H.; Ung, P.M.; Zajkowski, J.; Garneau-Tsodikova, S.; Sherman, D.H. Automated genome mining for natural products. BMC Bioinf. 2009, 10, 1–10. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Johnston, C.W.; Edgar, R.E.; Dejong, C.A.; Merwin, N.J.; Rees, P.N.; Magarvey, N.A. Genomic charting of ribosomally synthesized natural product chemical space facilitates targeted mining. Proc. Natl. Acad. Sci. USA 2016, 113, E6343–E6351. [Google Scholar] [CrossRef]
- Skinnider, M.A.; Johnston, C.W.; Gunabalasingam, M.; Merwin, N.J.; Kieliszek, A.M.; MacLellan, R.J.; Li, H.; Ranieri, M.R.; Webster, A.L.; Cao, M.P.; et al. Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Blin, K.; Medema, M.H.; Kazempour, D.; Fischbach, M.A.; Breitling, R.; Takano, E.; Weber, T. antiSMASH 2.0—A versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41, W204–W212. [Google Scholar] [CrossRef]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; De Los Santos, E.L.; Kim, H.U.; Nave, M.; et al. antiSMASH 4.0—Improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Röttig, M.; Medema, M.H.; Blin, K.; Weber, T.; Rausch, C.; Kohlbacher, O. NRPSpredictor2—A web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011, 39, W362–W367. [Google Scholar] [CrossRef] [PubMed]
- Chevrette, M.G.; Aicheler, F.; Kohlbacher, O.; Currie, C.R.; Medema, M.H. SANDPUMA: Ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 2017, 33, 3202–3210. [Google Scholar] [CrossRef]
- Süssmuth, R.D.; Mainz, A. Nonribosomal peptide synthesis—Principles and prospects. Angew. Chem. Int. Ed. 2017, 56, 3770–3821. [Google Scholar] [CrossRef]
- Juguet, M.; Lautru, S.; Francou, F.X.; Nezbedová, Š.; Leblond, P.; Gondry, M.; Pernodet, J.L. An iterative nonribosomal peptide synthetase assembles the pyrrole-amide antibiotic congocidine in Streptomyces ambofaciens. Chem. Biol. 2009, 16, 421–431. [Google Scholar] [CrossRef]
- Sieber, S.A.; Marahiel, M.A. Learning from Nature’s Drug Factories: Nonribosomal Synthesis of Macrocyclic Peptides. J. Bacteriol. 2003, 185, 7036–7043. [Google Scholar] [CrossRef]
- Agrawal, P.; Mohanty, D. A machine learning-based method for prediction of macrocyclization patterns of polyketides and non-ribosomal peptides. Bioinformatics 2021, 37, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Paalvast, Y.; Nguyen, D.D.; Melnik, A.; Dorrestein, P.C.; Takano, E.; Breitling, R. Pep2Path: Automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput. Biol. 2014, 10, e1003822. [Google Scholar] [CrossRef]
- Mohimani, H.; Liu, W.T.; Kersten, R.D.; Moore, B.S.; Dorrestein, P.C.; Pevzner, P.A. NRPquest: Coupling mass spectrometry and genome mining for nonribosomal peptide discovery. J. Nat. Prod. 2014, 77, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Behsaz, B.; Bode, E.; Gurevich, A.; Shi, Y.N.; Grundmann, F.; Acharya, D.; Caraballo-Rodríguez, A.M.; Bouslimani, A.; Panitchpakdi, M.; Linck, A.; et al. Integrating genomics and metabolomics for scalable non-ribosomal peptide discovery. Nat. Commun. 2021, 12, 1–17. [Google Scholar]
- Zierep, P.F.; Ceci, A.T.; Dobrusin, I.; Rockwell-Kollmann, S.C.; Günther, S. SeMPI 2.0—A Web Server for PKS and NRPS Predictions Combined with Metabolite Screening in Natural Product Databases. Metabolites 2021, 11, 13. [Google Scholar] [CrossRef]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; De Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.; Van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kautsar, S.A.; Medema, M.H.; Weber, T. The antiSMASH database version 3: Increased taxonomic coverage and new query features for modular enzymes. Nucleic Acids Res. 2021, 49, D639–D643. [Google Scholar] [CrossRef]
- Flissi, A.; Ricart, E.; Campart, C.; Chevalier, M.; Dufresne, Y.; Michalik, J.; Jacques, P.; Flahaut, C.; Lisacek, F.; Leclère, V.; et al. Norine: Update of the nonribosomal peptide resource. Nucleic Acids Res. 2020, 48, D465–D469. [Google Scholar]
- Moumbock, A.F.; Gao, M.; Qaseem, A.; Li, J.; Kirchner, P.A.; Ndingkokhar, B.; Bekono, B.D.; Simoben, C.V.; Babiaka, S.B.; Malange, Y.I.; et al. StreptomeDB 3.0: An updated compendium of streptomycetes natural products. Nucleic Acids Res. 2021, 49, D600–D604. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Ricart, E.; Leclère, V.; Flissi, A.; Mueller, M.; Pupin, M.; Lisacek, F. rBAN: Retro-biosynthetic analysis of nonribosomal peptides. J. Cheminf. 2019, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Mikheenko, A.; Shlemov, A.; Korobeynikov, A.; Mohimani, H.; Pevzner, P.A. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat. Microbiol. 2018, 3, 319. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, J.A.; Jacob, G.; Singh, A.L.; Aniebok, V.; Balunas, M.J.; Bunsko, D.; Neto, F.C.; Castaño-Espriu, L.; Chang, C.; Clark, T.N.; et al. The natural products atlas: An open access knowledge base for microbial natural products discovery. ACS Central Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef]
- Agrawal, P.; Khater, S.; Gupta, M.; Sain, N.; Mohanty, D. RiPPMiner: A bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res. 2017, 45, W80–W88. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kim, E.; Shin, Y.H.; Kim, T.H.; Byun, W.S.; Cui, J.; Du, Y.E.; Lim, H.J.; Song, M.C.; Kwon, A.S.; Kang, S.H.; et al. Characterization of the ohmyungsamycin biosynthetic pathway and generation of derivatives with improved antituberculosis activity. Biomolecules 2019, 9, 672. [Google Scholar] [CrossRef]
- Ganley, J.G.; Carr, G.; Ioerger, T.R.; Sacchettini, J.C.; Clardy, J.; Derbyshire, E.R. Discovery of antimicrobial lipodepsipeptides produced by a Serratia sp. within mosquito microbiomes. ChemBioChem 2018, 19, 1590–1594. [Google Scholar] [CrossRef]
- Um, S.; Choi, T.J.; Kim, H.; Kim, B.Y.; Kim, S.H.; Lee, S.K.; Oh, K.B.; Shin, J.; Oh, D.C. Ohmyungsamycins A and B: Cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J. Organic Chem. 2013, 78, 12321–12329. [Google Scholar] [CrossRef]
- Namikoshi, M.; Sun, F.; Choi, B.W.; Rinehart, K.L.; Carmichael, W.W.; Evans, W.R.; Beasley, V.R. Seven more microcystins from homer lake cells: Application of the general method for structure assignment of peptides containing. Alpha.,. Beta.-dehydroamino acid unit(s). J. Organic Chem. 1995, 60, 3671–3679. [Google Scholar] [CrossRef]
- Kjaerulff, L.; Nielsen, A.; Mansson, M.; Gram, L.; Larsen, T.O.; Ingmer, H.; Gotfredsen, C.H. Identification of four new agr quorum sensing-interfering cyclodepsipeptides from a marine Photobacterium. Mar. Drugs 2013, 11, 5051–5062. [Google Scholar] [CrossRef]
- Machado, H.; Giubergia, S.; Mateiu, R.V.; Gram, L. Photobacterium galatheae sp. nov., a bioactive bacterium isolated from a mussel in the Solomon Sea. Int. J. Syst. Evol. Microbiol. 2015, 65, 4503–4507. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. [Google Scholar] [CrossRef] [PubMed]
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 2017, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Mohimani, H.; Gurevich, A.; Shlemov, A.; Mikheenko, A.; Korobeynikov, A.; Cao, L.; Shcherbin, E.; Nothias, L.F.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Xu, Y.; Kersten, R.D.; Nam, S.J.; Lu, L.; Al-Suwailem, A.M.; Zheng, H.; Fenical, W.; Dorrestein, P.C.; Moore, B.S.; Qian, P.Y. Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 2012, 134, 8625–8632. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef]
- Rausch, C.; Weber, T.; Kohlbacher, O.; Wohlleben, W.; Huson, D.H. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs). Nucleic Acids Res. 2005, 33, 5799–5808. [Google Scholar] [CrossRef]
- Magarvey, N.A.; Haltli, B.; He, M.; Greenstein, M.; Hucul, J.A. Biosynthetic pathway for mannopeptimycins, lipoglycopeptide antibiotics active against drug-resistant gram-positive pathogens. Antimicrob. Agents Chemother. 2006, 50, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Orozco, R.; Wijeratne, E.K.; Espinosa-Artiles, P.; Gunatilaka, A.L.; Stock, S.P.; Molnár, I. Biosynthesis of the cyclooligomer depsipeptide bassianolide, an insecticidal virulence factor of Beauveria bassiana. Fungal Genet. Biol. 2009, 46, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Stachelhaus, T. Selective interaction between nonribosomal peptide synthetases is facilitated by short communication-mediating domains. Proc. Natl. Acad. Sci. USA 2004, 101, 15585–15590. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 30 September 2021).
- Hagberg, A.; Swart, P.; Chult, D.S. Exploring Network Structure, Dynamics, and Function Using NetworkX; Technical Report; Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2008. [Google Scholar]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinf. 2011, 12, 1–10. [Google Scholar] [CrossRef]
- Chen, X.H.; Koumoutsi, A.; Scholz, R.; Borriss, R. More than anticipated–production of antibiotics and other secondary metabolites by Bacillus amyloliquefaciens FZB42. J. Mol. Microbiol. Biotechnol. 2009, 16, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Tambadou, F.; Caradec, T.; Gagez, A.L.; Bonnet, A.; Sopéna, V.; Bridiau, N.; Thiéry, V.; Didelot, S.; Barthélémy, C.; Chevrot, R. Characterization of the colistin (polymyxin E1 and E2) biosynthetic gene cluster. Arch. Microbiol. 2015, 197, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Seyedsayamdost, M.R.; Traxler, M.F.; Zheng, S.L.; Kolter, R.; Clardy, J. Structure and biosynthesis of amychelin, an unusual mixed-ligand siderophore from Amycolatopsis sp. AA4. J. Am. Chem. Soc. 2011, 133, 11434–11437. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]




| # Genomes | Structure Parsing | Genome Mining | Matching | The Full Pipeline | ||||
|---|---|---|---|---|---|---|---|---|
| GRAPE | rBAN | PRISM | antiSMASH | GARLIC | Nerpa | GARLIC | Nerpa | |
| Running Time (d-h:m) | ||||||||
| 100 | 3:41 | 0:17 | 3:52 ± 1:41 | 0:05 ± 0:01 | 13:00 ± 1:09 | 0:57 ± 1:24 | 20:33 ± 2:41 | 1:19 ± 1:25 |
| 13,399 | ∼21-14:08 | 23:36 | ∼72-14:00 | 1-16:14 | ∼94-04:08 | 2-16:07 | ||
| Peak RAM Consumption (GB) | ||||||||
| 100 | 16.3 | 13.1 | 40.2 ± 1.2 | 3.6 ± 0.4 | 2.5 ± 0.9 | 0.6 ± 0.4 | 40.2 ± 1.2 | 13.1 |
| 13,399 | ∼40.2 | 4.8 | ∼2.5 | 18.6 | ∼40.2 | 18.6 | ||
| ID pNRPdb | Compound | Producer | ID GenBank | Species | Score | Known Analog |
|---|---|---|---|---|---|---|
| NPA011095 | Ohmyungsamycin A | Streptomyces sp. [41] | GCA_013364095.1 | S. harbinensis | 27.31 | QGA70148.1 (ohmA) |
| NPA014983 | Microcystin-LR’ | Microcystis sp. [42] | GCA_000010625.1 | M. aeruginosa | 13.88 | BGC0001017 (mcyA,B,C,E) |
| NPA002702 | Ngercheumicin F | Photobacterium sp. [43] | GCA_000695255.1 | P. galatheae | 12.76 | — |
| NPA024438 | Stephensiolide F | Serratia sp. [40] | GCA_017309605.1 | S. ureilytica | 11.69 | ATD12179.1 (sphA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunyavskaya, O.; Tagirdzhanov, A.M.; Caraballo-Rodríguez, A.M.; Nothias, L.-F.; Dorrestein, P.C.; Korobeynikov, A.; Mohimani, H.; Gurevich, A. Nerpa: A Tool for Discovering Biosynthetic Gene Clusters of Bacterial Nonribosomal Peptides. Metabolites 2021, 11, 693. https://doi.org/10.3390/metabo11100693
Kunyavskaya O, Tagirdzhanov AM, Caraballo-Rodríguez AM, Nothias L-F, Dorrestein PC, Korobeynikov A, Mohimani H, Gurevich A. Nerpa: A Tool for Discovering Biosynthetic Gene Clusters of Bacterial Nonribosomal Peptides. Metabolites. 2021; 11(10):693. https://doi.org/10.3390/metabo11100693
Chicago/Turabian StyleKunyavskaya, Olga, Azat M. Tagirdzhanov, Andrés Mauricio Caraballo-Rodríguez, Louis-Félix Nothias, Pieter C. Dorrestein, Anton Korobeynikov, Hosein Mohimani, and Alexey Gurevich. 2021. "Nerpa: A Tool for Discovering Biosynthetic Gene Clusters of Bacterial Nonribosomal Peptides" Metabolites 11, no. 10: 693. https://doi.org/10.3390/metabo11100693
APA StyleKunyavskaya, O., Tagirdzhanov, A. M., Caraballo-Rodríguez, A. M., Nothias, L.-F., Dorrestein, P. C., Korobeynikov, A., Mohimani, H., & Gurevich, A. (2021). Nerpa: A Tool for Discovering Biosynthetic Gene Clusters of Bacterial Nonribosomal Peptides. Metabolites, 11(10), 693. https://doi.org/10.3390/metabo11100693