Using Out-of-Batch Reference Populations to Improve Untargeted Metabolomics for Screening Inborn Errors of Metabolism

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
2.1. Data and Batch Characteristics
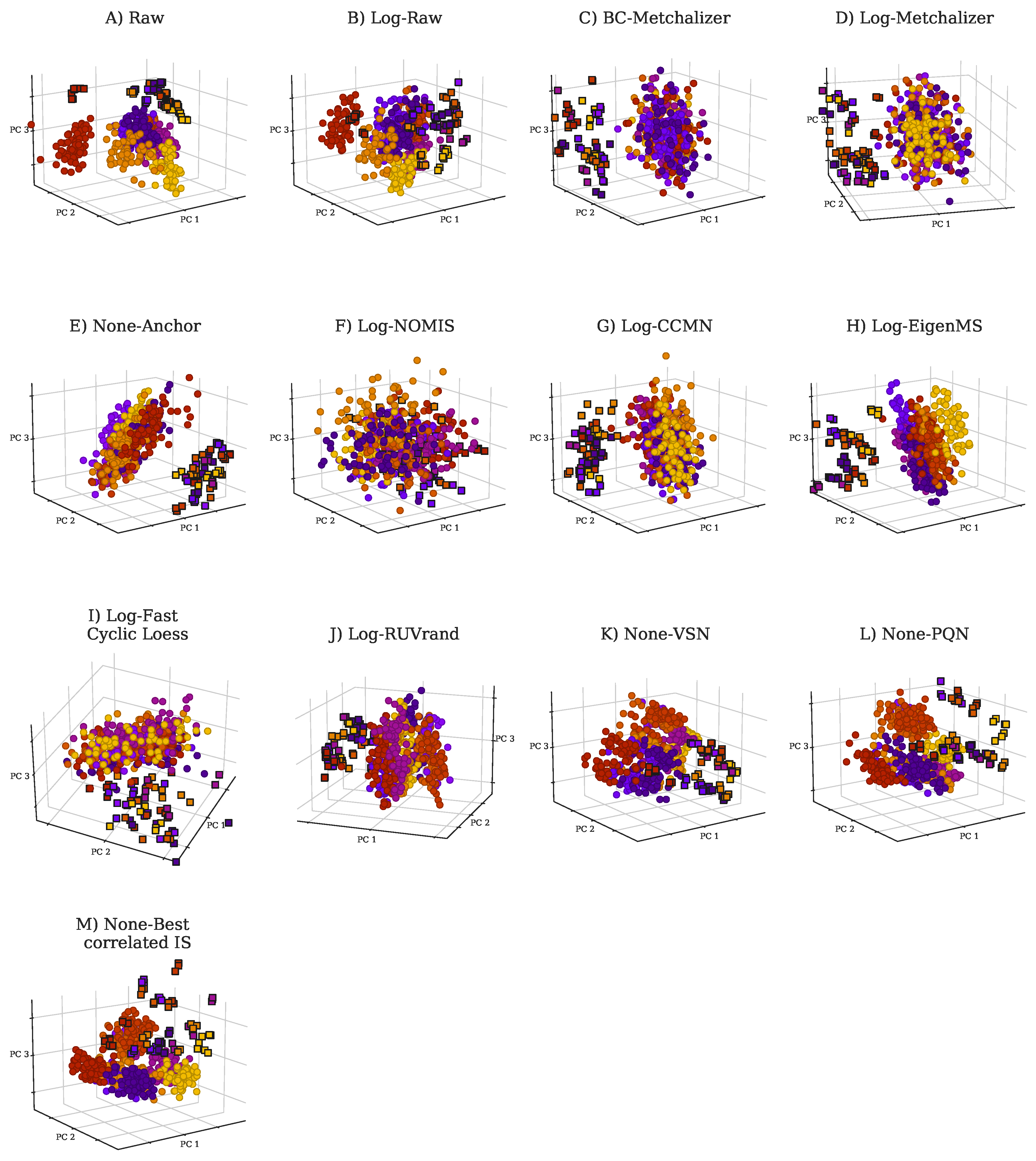
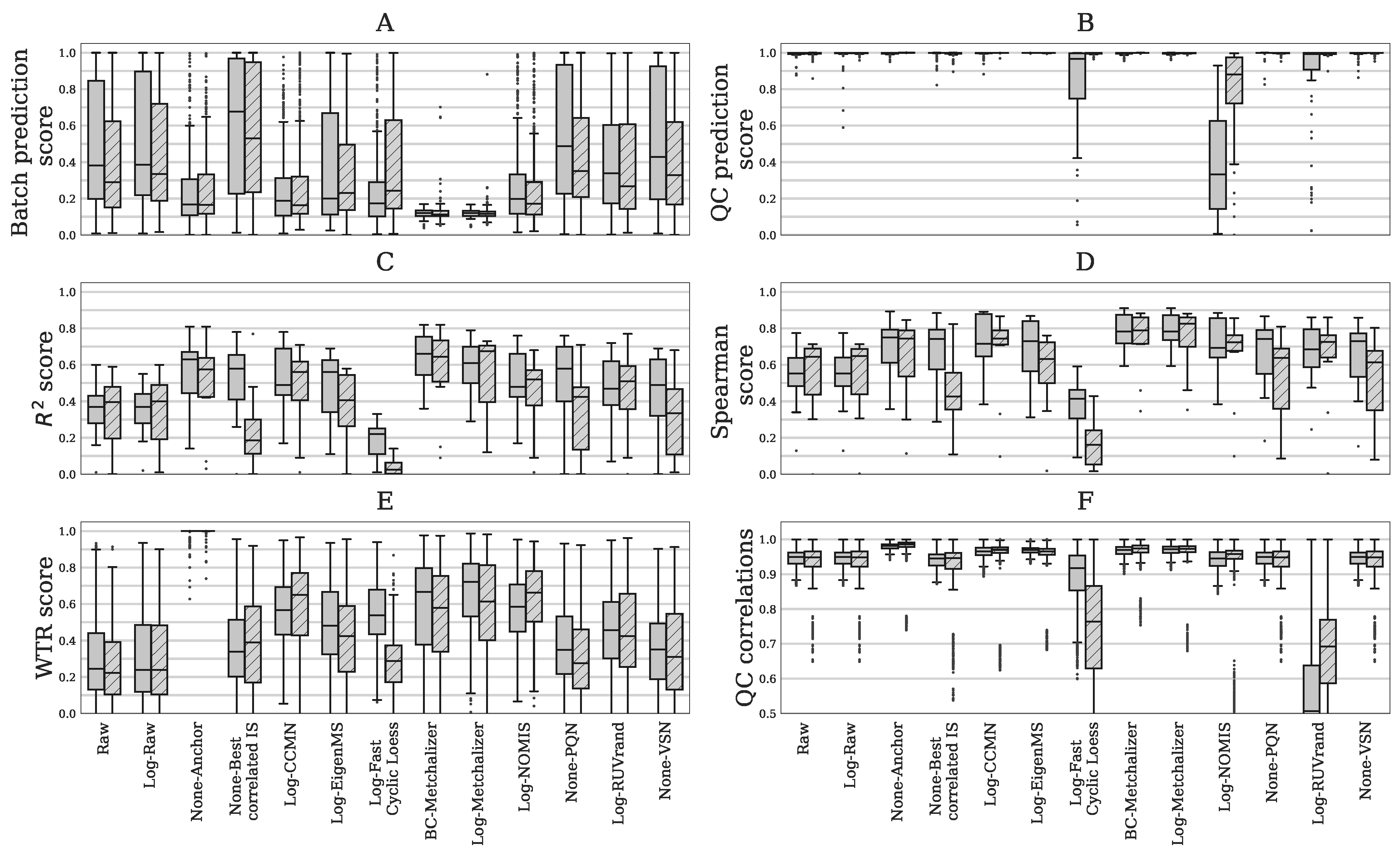
2.2. Comparing Normalization Methods
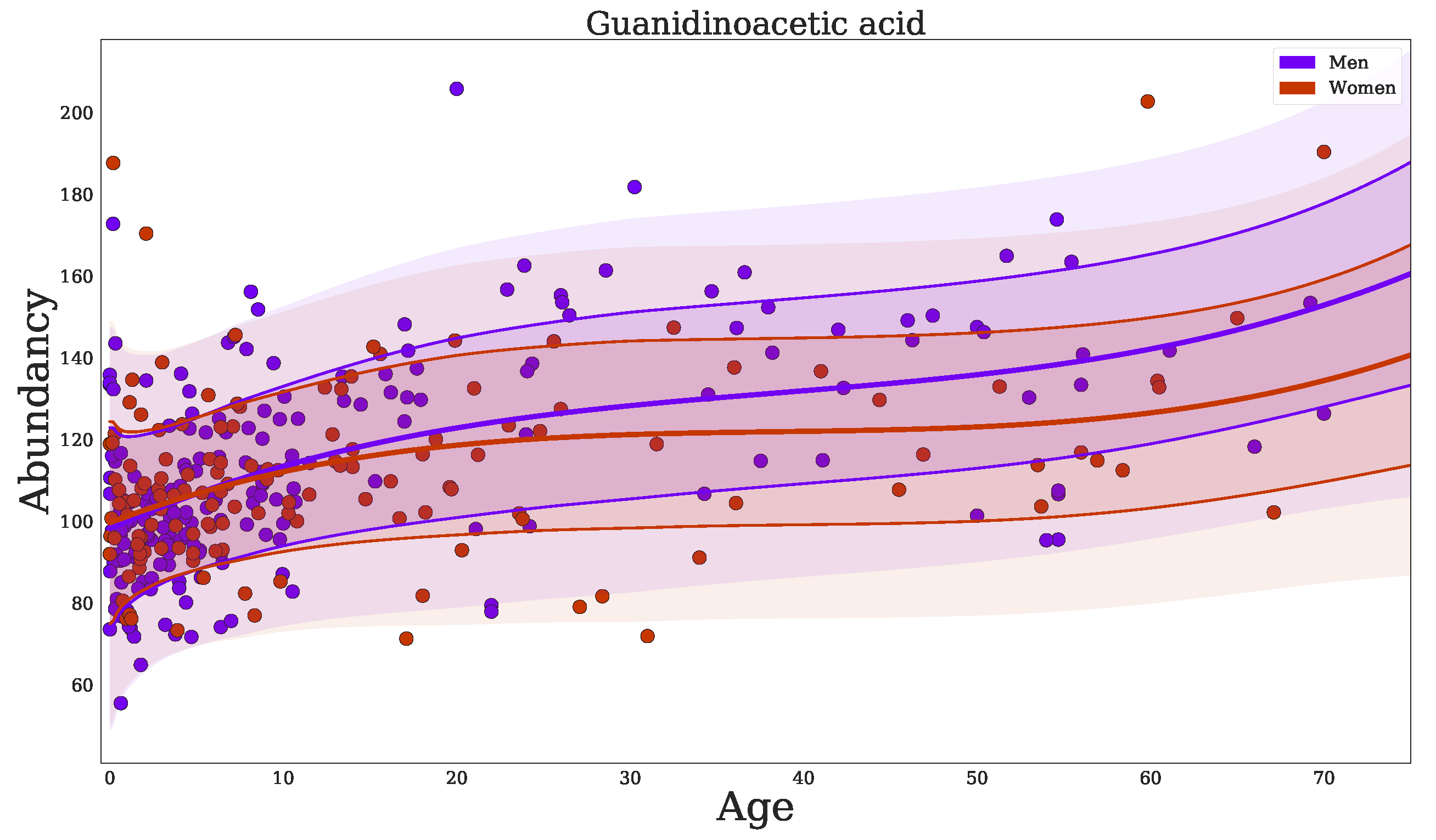
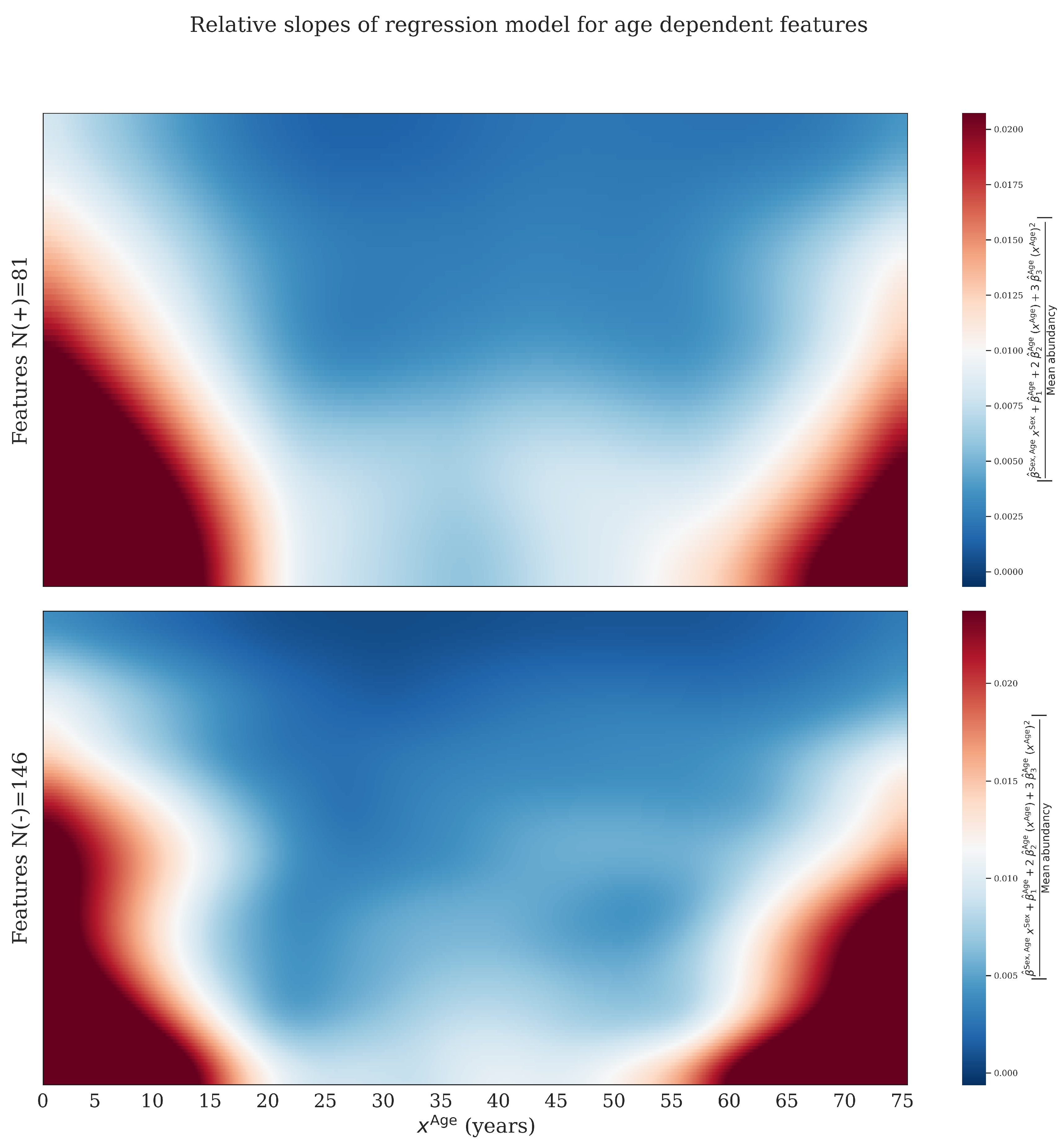
2.3. Confounder Effects of Age and Sex
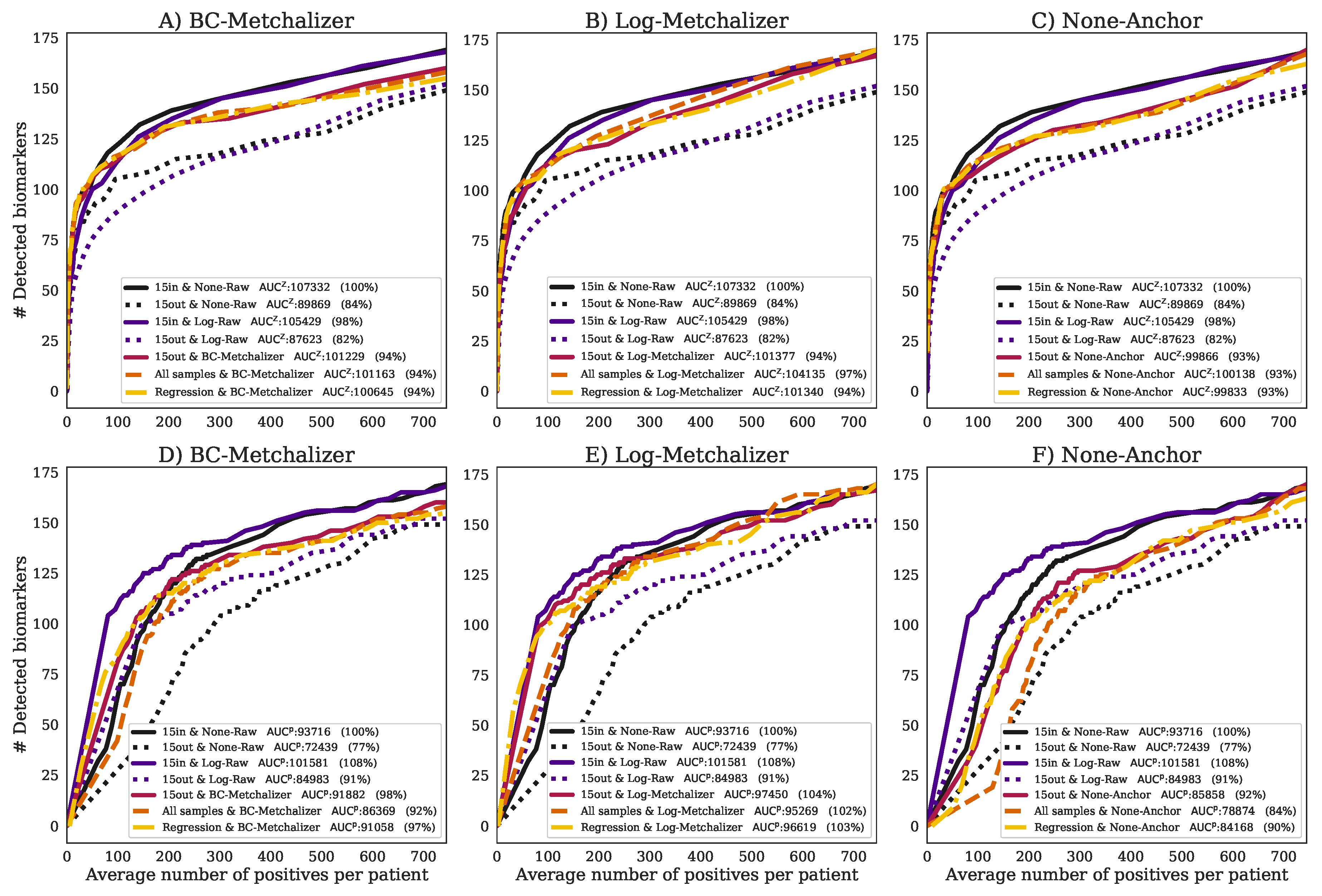
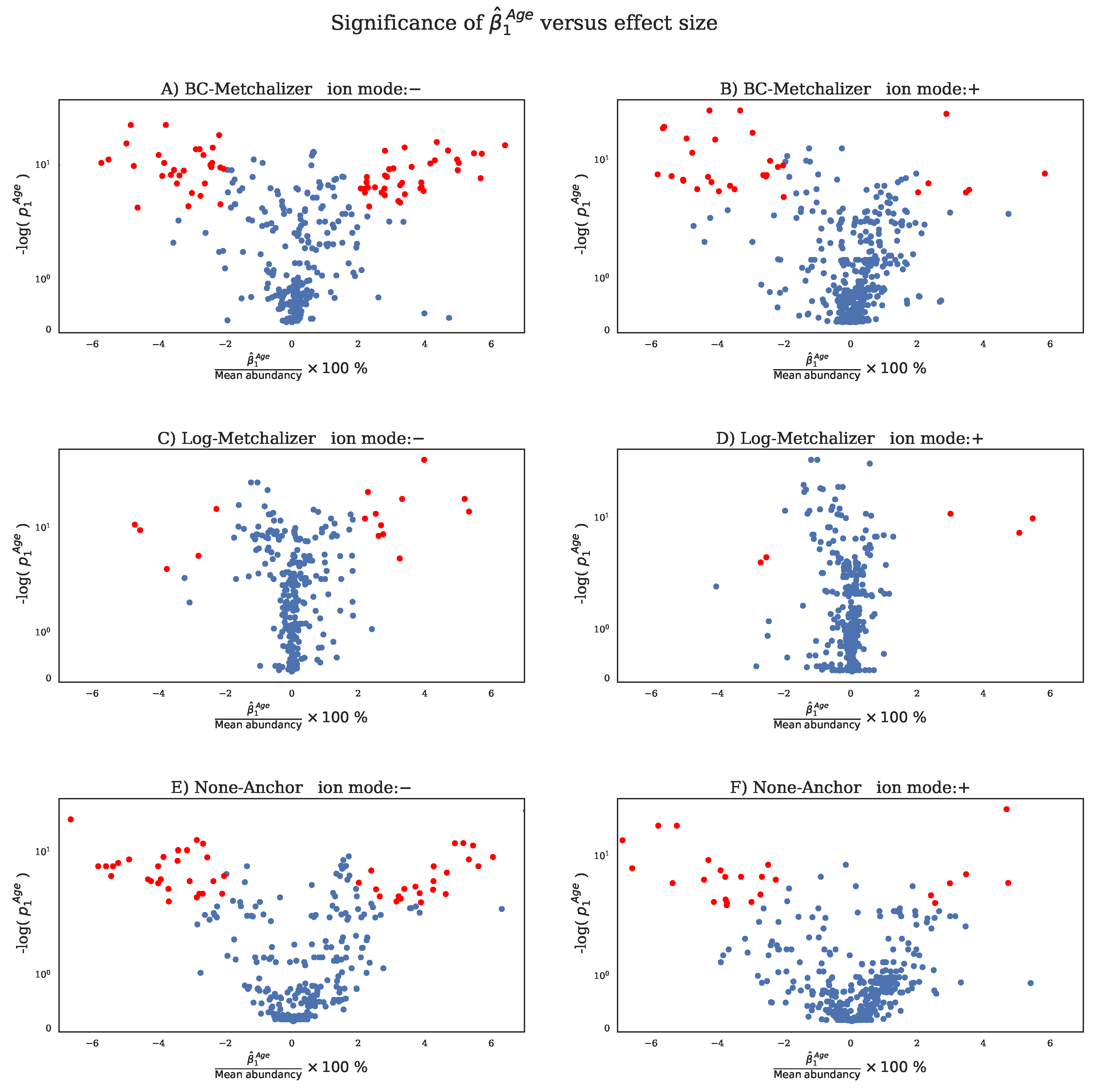
2.4. Detection of the Expected IEM Biomarkers
3. Discussion
4. Materials and Methods
4.1. Untargeted Metabolomics Datasets
4.2. Data Processing
- When features were annotated in reference batch and the batch being merged, these features were pooled to the merged dataset.
- When MS/MS spectra were present for a potential matching pair of features, the cosine similarity metric was calculated and it had to be >0.8.
- The retention time difference in percentage was calculated between potential matches, and it had to be <3%.
- Progenesis QI determined per feature an isotope distribution and we required sufficient overlap of these distributions between potential matching pairs. This was determined by calculating a difference in the percentage between each bin of this distribution. The maximum difference of these bins had to be <25%.
- Despite the batch effects and potential biological differences between batches, we expected the within-batch median of the (raw) abundancies for matching features to be at least similar. We calculated the differences between these medians in percentages, and required that this difference was <300%.
- When neutral masses were known for the matching pair, but not the MS/MS spectra, the ppm-error had to be <1.
- When m/z-values were known for the matching pair, but not the MS/MS spectra and neutral masses, the ppm-error of between the m/z-values had to be <1.
4.3. Quantitative Evaluation Set
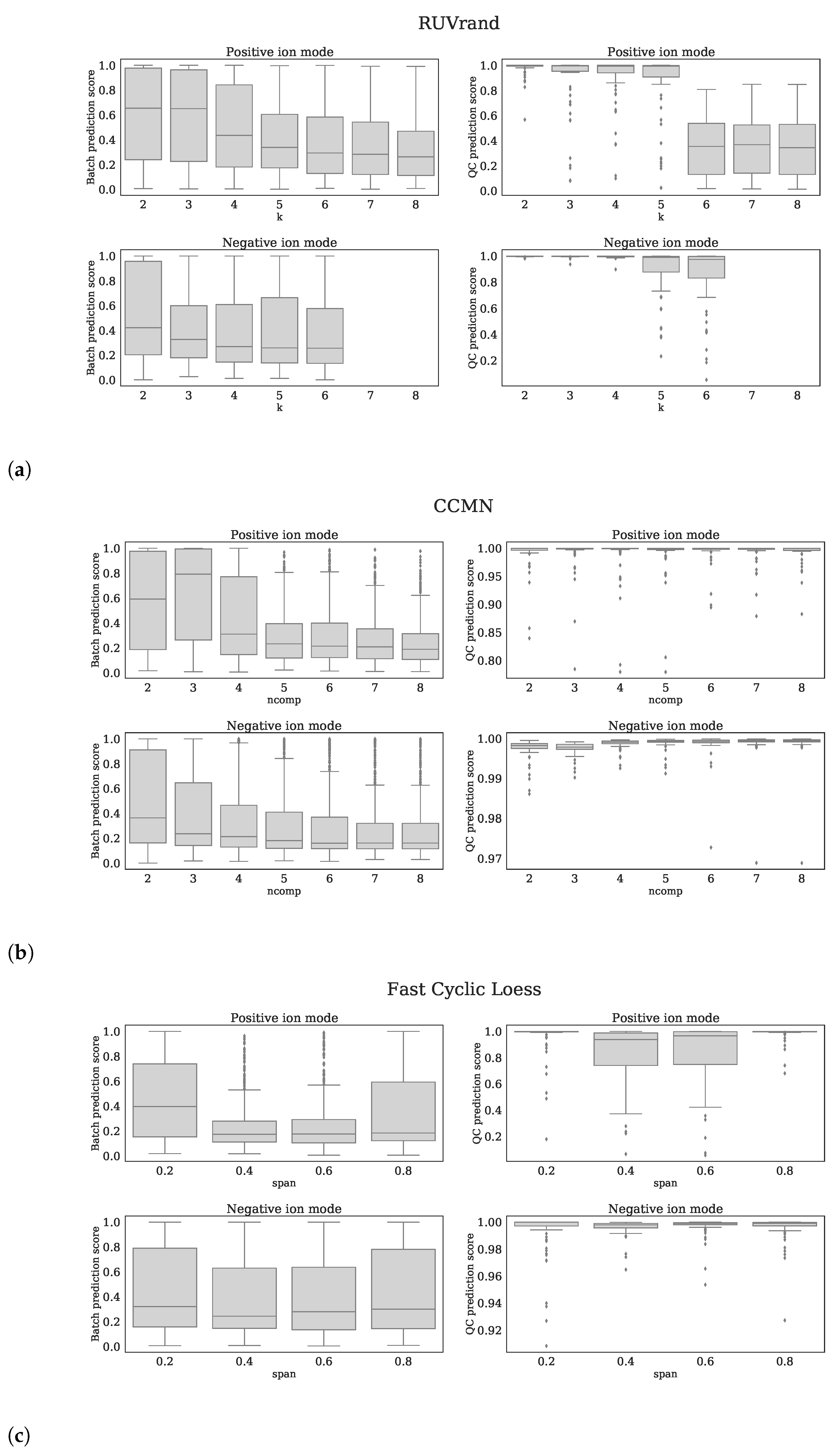
4.4. Normalization Methods
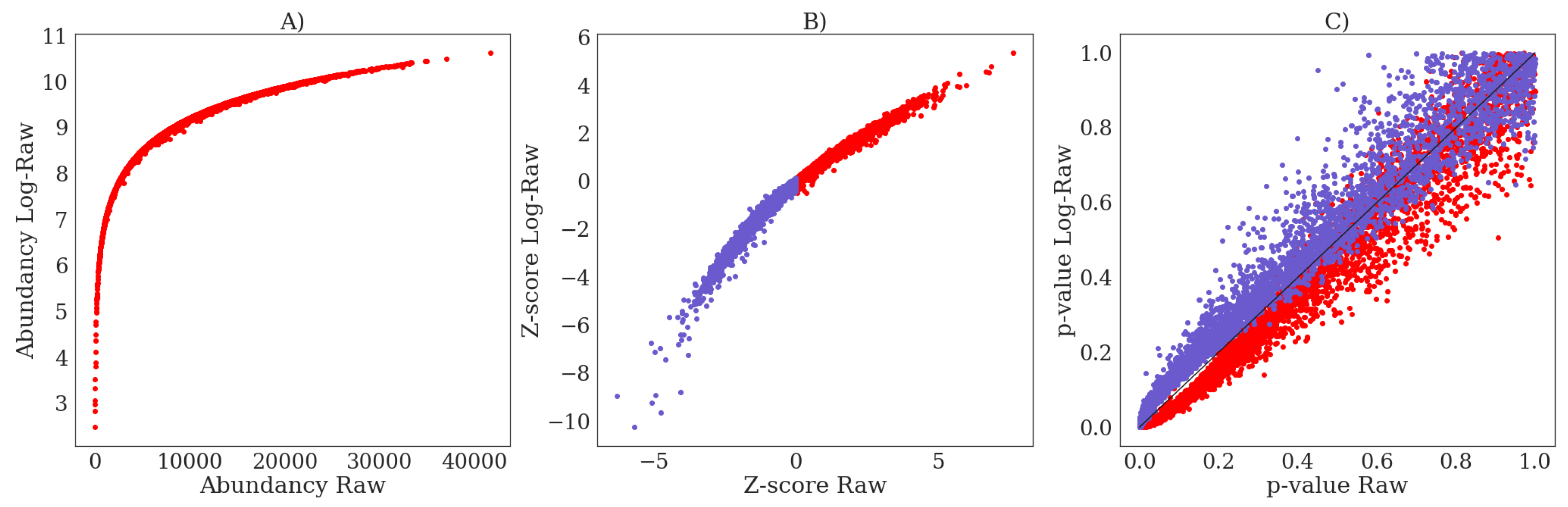
4.4.1. Initial Transformations
4.4.2. Normalization by Metchalizer
4.4.3. Normalization by Best Correlated IS
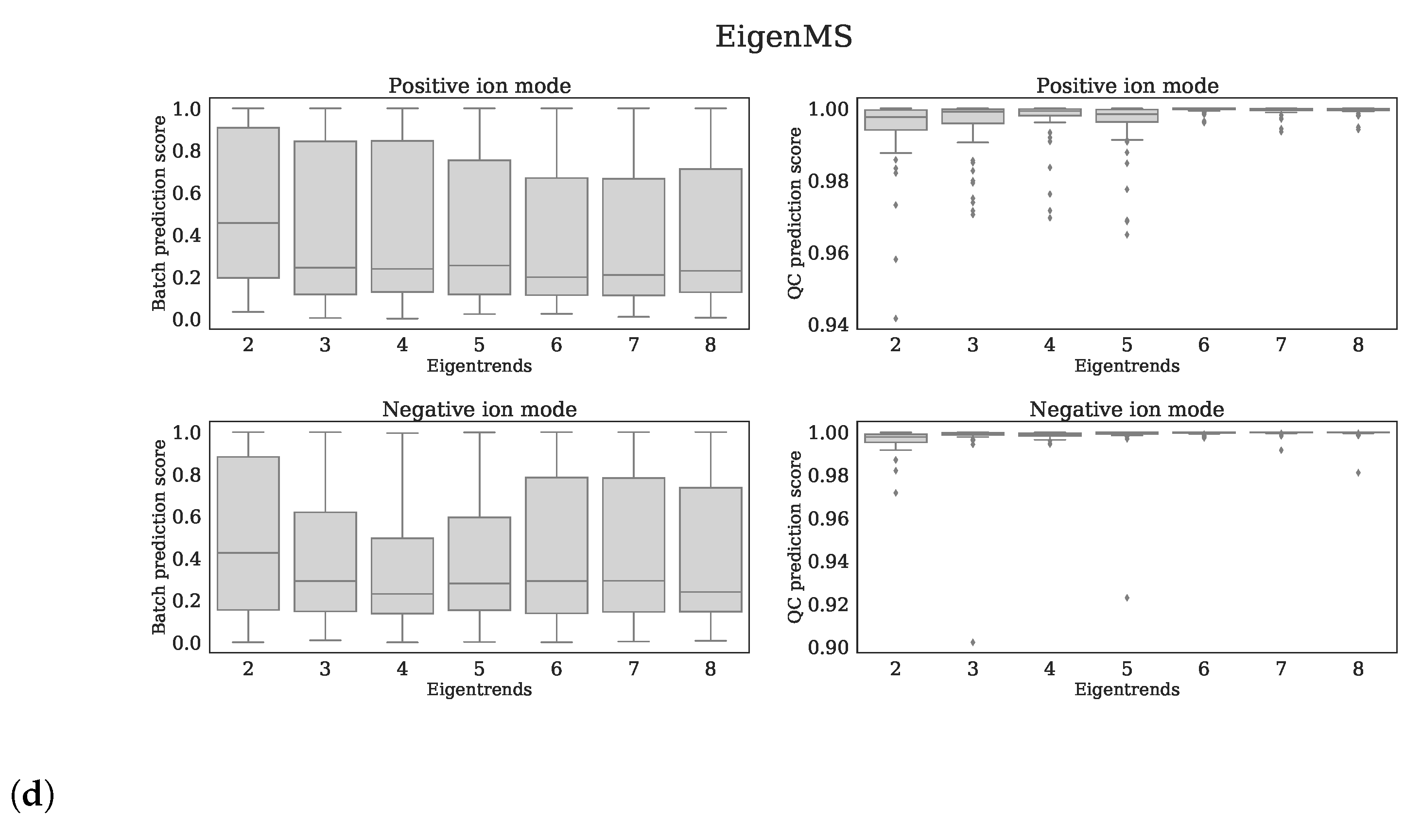
4.4.4. Normalization Methods from Literature
- Anchor [6]: Anchor assumes a linear response between the features in the anchor samples and samples in the batch. An anchor sample is a fixed sample, which is analyzed in all nine batches, and it was included more than four times in each batch. Normalization was performed per batch by dividing each feature by the average of the anchor samples for that same feature per batch. In this study, we used our QC samples as the anchor samples.
- CCMN [15]: we used function normFit from the crmn R package with input argument ‘crmn’. As a design matrix, we chose QC samples versus human plasma’s.
- EigenMS [16]: QC samples and human plasma samples were treated as two different groups.
- Fast Cyclic Loess [17]: we used the normalize CyclicLoess function from the limma R package while using the method ‘fast’ and iterations=100.
- NOMIS [18]: we used the function normFit from the crmn R package with input argument ‘nomis’.
- PQN [19]: PQN was implemented, as described by Filzmoser et al. The reference spectrum was given by the median of every feature j.
- RUV [20]: we used the function RUVRand from the MetNorm R package.
- VSN [21]: we used the vsn R package while using the vsn2 function.
4.4.5. Evaluation of Normalization Methods
- WTR score: the WTR score (Within variance Total variance Ratio) calculates the ratio between the ‘overall’ within-batch variance and the total variance from the QC samples:where is the variance of all nine batch averages for metabolite j in the QC samples, and the ‘overall’ variance based on all QC samples. The WTR score is between 0 and 1. Because we would like batch averages to be similar for the QC samples (resulting in approaching zero), we are interested in WTR scores close to one. Note that the coefficient of variation (CV) was considered to be an inadequate metric, as a simple log-transformation of the data already results in a decreased CV. Because the WTR score considers a ratio between two standard deviations, this metric is less sensitive to such initial data transformations.
- QC correlations: for all QC samples, the Spearman correlations were calculated on the (normalized) abundancies. Normalization should increase the resemblance of the QC samples among each other, therefore increasing the Spearman correlations. It is expected that the Spearman correlations decreases when variations other then techical variation are removed.
- Spearman score: for the set of 15 quantitatively measured metabolites, we calculated the Spearman correlation between their quantitative measurements and the normalized abundancies. The overall normalization performance could be judged based on the median Spearman score of these 15 scores, having scores . Higher values indicate better resemblance with the quantitative measurements.
- score: the between the quantitative measurements and the normalized abundancies of the 15 quantitatively measured metabolites. The overall performance could be judged from the median score, with scores of . Higher values indicate better (linear) fits with the quantitative measurements.
- QC prediction score: since the QC samples were different from the human plasma samples in terms of concentrations for several metabolites/features, we expect this difference to be observed in the first few principal components (PCs) of a Principal Component Analysis (PCA) analysis applied to all features (excl. standards). We fitted a logistic function while using the first four PCs as covariates and with class labels: ‘human plasma’ and ‘QC’. The fitted model returns per sample a probability of belonging either to the class ‘human plasma’ or ‘QC’. The probabilities for all samples are averaged into the QC prediction score. Increasing normalization performances should result in higher scores, as QC- and human plasma samples should be segregated. We used LogisticRegression from the Python package scikitlearn with parameters penalty=‘l1’, solver=‘saga’, multi_class=‘auto’, and max_iter=10,000 [22].
- Batch prediction score: increasing normalization performances should result in less batch clustering when examining the first few PCs of the PCA analysis (see QC prediction score). We fitted a logistic function for each batch versus all other eight batches while using the first four PCs as covariates and obtained the probability scores for all human plasma’s having the correct batch label. These scores were than averaged for all human plasma samples into a batch prediction scores . Scores that are closer to 1 indicate decreased normalization performances, since batch separation is (still) present.
4.4.6. Settings for Normalization Methods from Literature
4.5. Methods to Determine Metabolic Abberations
4.5.1. Outlier Removal
4.5.2. Z-Score Methods
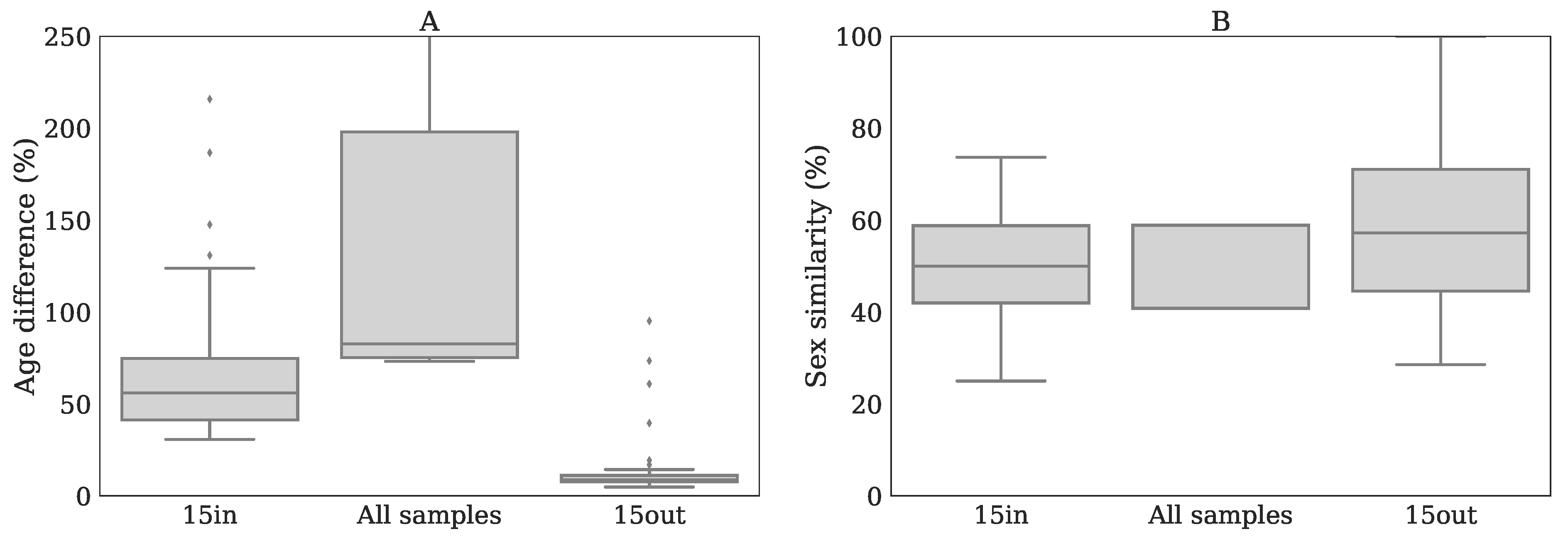
- 15in, best matching samples within batch: the Z-scores were calculated by selecting 15 samples originating from the same batch that were matched with the patient based on age and sex, as described in Bonte et al. [5].
- 15out, best matching samples from other batches: the Z-scores were calculated similarly as in method 15in while using 15 out-of-batch samples. Note that since there are more out-of-batch samples than within-batch samples the age and sex matching can be done more accurate for 15out than for 15in.
- All samples: this method used all available reference samples from all nine batches, including within-batch controls, for Z-score calculation, thereby ignoring age- and sex matching.
- Regression: we fitted a linear model on all available reference samples excluding outliers that were first removed based on a within-batch . This Z-score is different from other Z-scores mentioned in this study, and it is only used to remove outliers. The regression model is given by:where is the predicted (normalized) abundancy of feature j for sample i, is an intercept. , (interaction) and indicate slopes. P is the degree of the polynomial used for regression on age and set to in this study. is 1 for women and 0 for men. is the estimated error. The latter expression is the model in vector notation with .
4.5.3. Final Z-Scores
4.5.4. p-Values from Welch’s T-Test
4.6. Detection of the Expected IEM Biomarkers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| ROC | Receiver operating characteristic |
| IEM | Inborn error of metabolism |
| CV | Coefficient of variation |
| QC | Quality control |
| PCA | Principle component analysis |
| PC | Principle component |
| UHPLC | Ultra-high performance liquid chromatography |
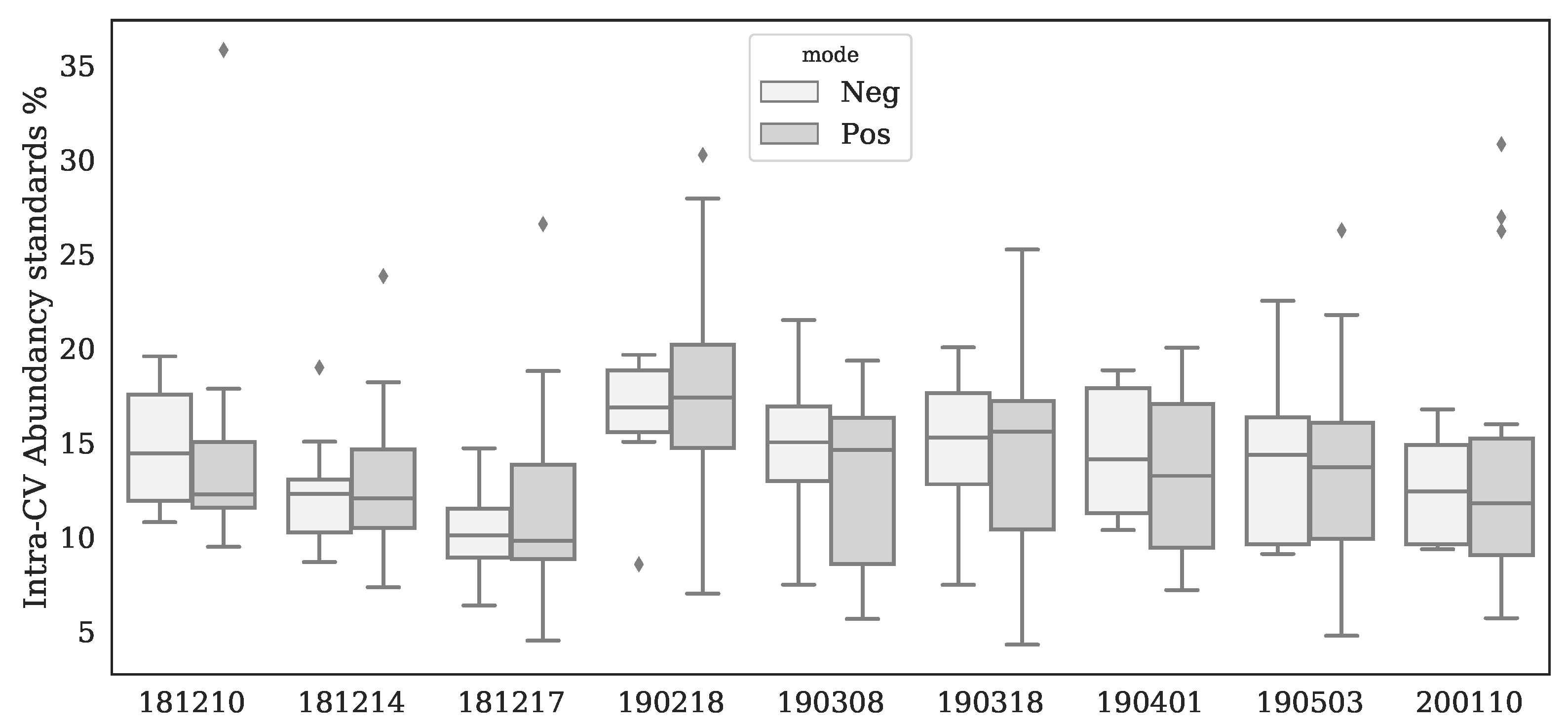
Appendix A. Variations of Standards


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standard | Ion Mode | Inter-Batch CV (%) |
|---|---|---|
| 3,3-dimethylglutaric acid | − | 23 |
| 3,3-dimethylglutaric acid | + | 72 |
| 5-bromotryptophane | − | 18 |
| 5-bromotryptophane | + | 21 |
| Acetylcarnitine D2 | + | 31 |
| Carnitine D3 | + | 23 |
| Glycochenodeoxycholic Acid D4 | − | 20 |
| Hexadecanoylcarnitine D3 | + | 31 |
| Hexanoylcarnitine D3 | + | 28 |
| Isoleucine D10 | − | 40 |
| Isoleucine D10 | + | 27 |
| Methylmalonic Acid D3 | − | 52 |
| Ornithine D6 | + | 26 |
| Phenylalanine D5 | − | 35 |
| Phenylalanine D5 | + | 29 |
| Tetradecanoylcarnitine D3 | + | 27 |
| Thymidine 13C | − | 18 |
| Thymidine 13C | + | 51 |
| Tyrosine D4 | − | 27 |
| Tyrosine D4 | + | 27 |
| Uracil-1,3N15 | − | 18 |
| Uracil-1,3N15 | + | 19 |
| Uridine D2 | − | 27 |
| Uridine D2 | + | 58 |
| Valine D8 | + | 41 |
Appendix B. Batch Effect Removal Performances
| Method | Ion Mode | QC Prediction Score | Score | Spearman Score | WTR Score | QC Correlations | 1—Batch Prediction Score | Mean |
|---|---|---|---|---|---|---|---|---|
| None-Anchor | + | 1.00 | 0.63 | 0.75 | 1.00 | 0.98 | 0.83 | 0.77 |
| None-Anchor | − | 1.00 | 0.57 | 0.74 | 1.00 | 0.99 | 0.83 | 0.76 |
| Log-Metchalizer | − | 1.00 | 0.68 | 0.83 | 0.61 | 0.97 | 0.88 | 0.73 |
| Log-Metchalizer | + | 1.00 | 0.61 | 0.78 | 0.72 | 0.97 | 0.88 | 0.73 |
| BC-Metchalizer | + | 1.00 | 0.66 | 0.78 | 0.67 | 0.97 | 0.88 | 0.73 |
| BC-Metchalizer | − | 1.00 | 0.64 | 0.79 | 0.58 | 0.97 | 0.89 | 0.71 |
| Log-CCMN | − | 1.00 | 0.56 | 0.74 | 0.65 | 0.97 | 0.84 | 0.70 |
| Log-NOMIS | − | 0.88 | 0.52 | 0.72 | 0.66 | 0.96 | 0.83 | 0.68 |
| Log-EigenMS | + | 1.00 | 0.56 | 0.73 | 0.48 | 0.97 | 0.80 | 0.68 |
| Log-CCMN | + | 1.00 | 0.49 | 0.72 | 0.57 | 0.97 | 0.81 | 0.68 |
| None-PQN | + | 1.00 | 0.58 | 0.74 | 0.35 | 0.95 | 0.51 | 0.66 |
| None-Best correlated IS | + | 1.00 | 0.58 | 0.74 | 0.34 | 0.95 | 0.32 | 0.66 |
| None-VSN | + | 1.00 | 0.49 | 0.73 | 0.35 | 0.95 | 0.57 | 0.65 |
| Log-EigenMS | − | 1.00 | 0.40 | 0.63 | 0.42 | 0.97 | 0.77 | 0.63 |
| Log-RUVrand | − | 1.00 | 0.51 | 0.73 | 0.43 | 0.69 | 0.73 | 0.62 |
| None-PQN | − | 1.00 | 0.42 | 0.64 | 0.28 | 0.95 | 0.65 | 0.61 |
| Log-Raw | − | 1.00 | 0.40 | 0.65 | 0.24 | 0.95 | 0.67 | 0.61 |
| Raw | − | 1.00 | 0.40 | 0.64 | 0.22 | 0.95 | 0.71 | 0.60 |
| None-VSN | − | 1.00 | 0.34 | 0.61 | 0.31 | 0.95 | 0.67 | 0.60 |
| Raw | + | 1.00 | 0.37 | 0.55 | 0.25 | 0.95 | 0.62 | 0.59 |
| Log-Raw | + | 1.00 | 0.37 | 0.55 | 0.24 | 0.95 | 0.61 | 0.59 |
| Log-RUVrand | + | 0.99 | 0.47 | 0.68 | 0.46 | 0.51 | 0.66 | 0.59 |
| Log-Fast Cyclic Loess | + | 0.97 | 0.22 | 0.41 | 0.54 | 0.92 | 0.83 | 0.58 |
| Log-NOMIS | + | 0.33 | 0.48 | 0.69 | 0.59 | 0.94 | 0.80 | 0.58 |
| None-Best correlated IS | − | 1.00 | 0.18 | 0.43 | 0.39 | 0.95 | 0.47 | 0.56 |
| Log-Fast Cyclic Loess | − | 1.00 | 0.02 | 0.16 | 0.29 | 0.76 | 0.76 | 0.46 |
Appendix C. Regression Analysis
| Metabolite | Ion Mode | Sign | ||||||
|---|---|---|---|---|---|---|---|---|
| 1-Methyladenosine (1) | + | down | 0 | 5.8 | 7.1 | 1.8 | 8.6 | 8.6 |
| 2-Aminoadipic acid | + | down | 0 | 7.3 | 9.8 | 1.6 | 8.6 | 8.6 |
| 2-Aminoadipic acid | - | down | 0 | 3.0 | 1.1 | 1.9 | 8.6 | 5.4 |
| 2-Ketoglutaric acid | - | down | 0 | 4.5 | 1.9 | 1.6 | 8.6 | 5.2 |
| 3-Methoxytyrosine | + | down | 0 | 4.1 | 4.3 | 2.6 | 8.6 | 8.6 |
| 3-Methylhistidine/1-Methylhistidine | + | up | 0 | 8.1 | 2.4 | 5.1 | 8.6 | 7.8 |
| 4-Pyridoxic acid | - | down | 0 | 7.1 | 1.6 | 4.2 | 8.6 | 6.4 |
| Acetoacetic acid/Succinic acid semialdehyde | + | down | 0 | 2.1 | 2.5 | 6.8 | 8.6 | 8.6 |
| Adenine | + | down | 0 | 1.1 | 1.1 | 3.3 | 8.6 | 8.6 |
| C2 Acetylcarnitine | + | down | 0 | 1.2 | 2.9 | 6.4 | 8.6 | 8.6 |
| C3 Propionylcarnitine | + | down | 0 | 7.7 | 1.4 | 5.2 | 8.6 | 8.3 |
| C8:1 Octenoylcarnitine | + | down | 0 | 4.8 | 3.7 | 6.9 | 8.6 | 8.6 |
| Chenodeoxycholic acid | - | up | 0 | 2.0 | 1.7 | 6.2 | 8.6 | 8.5 |
| Cholesterol | + | up | 0 | 3.2 | 2.9 | 5.4 | 8.6 | 8.6 |
| Citrulline | - | up | 0 | 3.7 | 7.0 | 1.5 | 8.6 | 2.9 |
| Creatine | - | down | 0 | 1.3 | 3.7 | 8.6 | 8.6 | 5.4 |
| Creatine | + | down | 0 | 6.0 | 4.2 | 8.8 | 8.6 | 8.3 |
| Creatinine | + | up | 0 | 5.1 | 4.2 | 2.7 | 8.6 | 7.8 |
| Dehydro-epiandrosteronsulfaat (dheas) | - | up | 4.4 | 6.0 | 6.2 | 5.2 | 8.6 | 5.4 |
| Dihydroxycholanoic acid + gly | - | down | 0 | 1.1 | 3.7 | 7.6 | 8.6 | 8.5 |
| Dimethylarginine (sdma + adma) | + | down | 0 | 1.9 | 3.3 | 4.6 | 8.6 | 8.6 |
| Dimethylarginine (sdma + adma) | - | down | 0 | 1.2 | 8.2 | 1.4 | 8.6 | 8.5 |
| Glycocholic acid | + | down | 0 | 7.7 | 4.6 | 8.8 | 8.6 | 8.6 |
| Glycocholic acid | - | down | 0 | 2.0 | 3.5 | 8.4 | 8.6 | 8.0 |
| Guanidinoacetic acid | + | up | 0 | 3.1 | 6.2 | 8.8 | 8.6 | 4.9 |
| Histidine | - | up | 0 | 1.3 | 7.1 | 2.6 | 8.6 | 8.5 |
| Homoarginine | + | up | 0 | 2.1 | 2.0 | 5.7 | 8.6 | 8.6 |
| Indoxylsulfuric acid | - | up | 0 | 4.3 | 7.2 | 2.0 | 8.6 | 8.5 |
| Kynurenin | + | down | 0 | 1.8 | 2.6 | 3.1 | 8.6 | 8.6 |
| L-Rhamnose | - | up | 0 | 7.2 | 1.8 | 4.4 | 8.6 | 4.8 |
| Lactic acid | - | down | 0 | 4.8 | 7.1 | 8.6 | 8.6 | 8.0 |
| N-Acetylaspartic acid | - | down | 0 | 7.9 | 3.5 | 4.7 | 8.6 | 7.9 |
| Pantothenic acid | - | down | 0 | 1.7 | 3.7 | 6.1 | 8.6 | 8.5 |
| Pantothenic acid | + | down | 0 | 4.0 | 1.2 | 3.5 | 8.6 | 8.6 |
| Phenylacetic acid | + | up | 0 | 1.8 | 1.2 | 2.6 | 8.6 | 8.6 |
| Pseudouridine | + | down | 0 | 9.4 | 1.8 | 1.6 | 8.6 | 8.6 |
| Raffinose/Hex3 | + | down | 0 | 7.2 | 4.2 | 1.6 | 8.6 | 8.6 |
| Raffinose/Hex3 | - | down | 0 | 3.0 | 1.6 | 4.8 | 8.6 | 8.5 |
| Ribose/Xylose/ Arabinose | - | down | 0 | 2.3 | 1.0 | 5.2 | 8.6 | 8.5 |
| Sialic acid | - | down | 0 | 4.1 | 1.6 | 8.6 | 8.6 | 6.5 |
| Sialic acid | + | down | 0 | 1.0 | 8.2 | 4.6 | 8.6 | 8.6 |
| Theophylline/ Paraxanthine | + | up | 0 | 8.3 | 8.5 | 8.8 | 8.6 | 5.8 |
| Trihydroxycholanoic acid + tau | - | down | 0 | 5.6 | 1.6 | 4.4 | 8.6 | 7.1 |
| Uric acid | + | up | 0 | 5.1 | 5.4 | 8.8 | 8.6 | 5.7 |
| Uric acid | - | up | 0 | 2.7 | 5.1 | 8.6 | 8.6 | 1.9 |
| Uridine | - | down | 0 | 3.6 | 3.3 | 6.6 | 8.6 | 8.5 |
| cis-Aconitic acid/trans-Aconitic acid | - | down | 0 | 4.1 | 6.8 | 7.4 | 8.6 | 8.5 |
| cis-Aconitic acid/trans-Aconitic acid | + | down | 0 | 1.4 | 2.5 | 6.9 | 8.6 | 8.6 |
| Metabolite | Mode | Sign | ||||||
|---|---|---|---|---|---|---|---|---|
| Guanidinoacetic acid | + | down | 0 | 3.1 | 6.2 | 8.8 | 8.6 | 4.9 |
| Ornithine | + | down | 0 | 8.4 | 7.1 | 8.8 | 8.6 | 4.9 |


| Method | Ion Mode | Percentage |
|---|---|---|
| BC-Metchalizer | − | 22.03 |
| BC-Metchalizer | + | 7.14 |
| Log-Metchalizer | − | 5.22 |
| Log-Metchalizer | + | 1.08 |
| None-Anchor | − | 17.10 |
| None-Anchor | + | 5.84 |
Appendix D. IEM Biomarkers
| Biomarker (Expected Z-Score Sign) (Ion Mode) | 15in&None-Raw | Regression&BC-Metchalizer | Regression&None-Anchor |
|---|---|---|---|
| Aminoacylase I deficiency N = 1 | |||
| N-Acetylarginine (up) (+) | 0.6 | 0.0 | 0.5 |
| N-Acetylglycine (up) (−) | 9.3 * | 4.2 * | 7.4 * |
| N-Acetylglycine (up) (+) | 5.3 * | 1.8 * | 4.6 * |
| Argininemia N = 1 | |||
| 4-Guanidinobutyric acid (up) (+) | 28.5 * | 11.4 * | 27.1 * |
| Arginine (up) (−) | 5.2 * | 4.0 * | 7.0 * |
| Arginine (up) (+) | 4.1 * | 3.1 * | 5.3 * |
| Glutamine + Glutamic acid/N-Methyl-D-Aspartic acid (up) (−) | 0.2 | 0.0 | 0.8 |
| Guanidinoacetic acid (down) (+) | 3.3 * | 2.1 * | 2.8 * |
| Homoarginine (up) (+) | 8.7 * | 10.7 * | 2.8 * |
| N-Acetylarginine (up) (+) | 117.2 * | 26.2 * | 95.7 * |
| Uridine (up) (−) | 3.0 * | 4.1 * | 4.1 * |
| Argininosuccinic aciduria N = 3 | |||
| Arginine (down) (−) | −0.9 *, −0.3, 0.8 * | −1.0 *, 0.2, 1.1 * | −1.0 *, 1.0, 0.9 * |
| Arginine (down) (+) | −1.0 *, −0.0, 1.0 * | −1.2 *, 0.3, 1.4 * | −1.3 *, 1.2 *, 1.3 * |
| Citrulline (up) (−) | 30.3 *, 20.3 *, 12.9 * | 9.8 *, 10.2 *, 8.6 * | 20.1 *, 17.4 *, 12.0 * |
| Citrulline (up) (+) | 19.9 *, 21.0 *, 11.2 * | 7.1 *, 10.8 *, 8.5 * | 14.4 *, 16.9 *, 10.6 * |
| Glutamine + Glutamic acid/N-Methyl-D-Aspartic acid (up) (−) | 1.9 *, 0.8 *, 0.9 * | 1.6 *, 1.2, 1.3 * | 1.2 *, 1.8 *, 1.7 * |
| Homocitrulline (up) (+) | 12.9 *, 2.1 *, 3.7 * | 5.3 *, 1.5 *, 1.9 * | 17.5 *, 2.3 *, 4.0 * |
| Uridine (up) (−) | 0.5, −0.4, 5.0 * | 0.3, −0.7 *, 4.7 * | 0.5 *, −0.6 *, 4.5 * |
| Beta-ketothiolase deficiency N = 2 | |||
| 2-Methylacetoacetic acid (up) (+) | 0.5, −0.9 * | 1.0, 0.7 | 0.4, 0.5 |
| Carbamoyl Phosphate Synthetase deficiency N = 2 | |||
| 2-Ketoglutaric acid (up) (−) | −1.2 *, −0.8 | −1.5 *, −1.1 | −0.8 *, −0.6 |
| Glutamine + Glutamic acid/N-Methyl-D-Aspartic acid (up) (−) | 3.5 *, −0.1 | 2.5 *, 0.2 | 4.1 *, 0.3 |
| Citrullinemia type I N = 1 | |||
| Arginine (down) (−) | 1.1 * | 1.1 | 0.0 |
| Arginine (down) (+) | 1.2 * | 1.1 * | 0.5 |
| Citrulline (up) (−) | 183.0 * | 54.3 * | 185.0 * |
| Citrulline (up) (+) | 104.9 * | 47.8 * | 112.5 * |
| Glutamine + Glutamic acid/N-Methyl-D-Aspartic acid (up) (−) | −0.5 | −1.4 | −1.0 * |
| Uridine (up) (−) | 1.4 * | 1.5 * | 0.7 |
| Glutamate formiminotransferase deficiency N = 1 | |||
| Formiminoglutamic acid (up) (+) | 1748.8 * | 135.6 * | 1060.0 * |
| Glutaric aciduria I N = 2 | |||
| C5DC Glutarylcarnitine (up) (+) | 360.3 *, 411.3 * | 26.9 *, 56.9 * | 309.5 *, 529.8 * |
| Glutaric acid (up) (+) | 0.4, 2.3 * | 0.3, 2.8 * | 1.1, 3.5 * |
| Glutaric aciduria II N = 2 | |||
| Adipic acid (1) (up) (−) | 1.5 *, 6.8 * | 1.2 *, 5.0 * | 1.4 *, 8.2 * |
| C10 Decanoylcarnitine (up) (+) | 114.9 *, 56.5 * | 23.5 *, 23.0 * | 65.3 *, 52.0 * |
| C12 Dodecanoylcarnitine (up) (+) | 0.9, 66.7 * | 0.5 *, 26.0 * | 1.2, 27.8 * |
| C14:1 Tetradecenoylcarnitine (up) (+) | 99.4, 68.7 * | 20.4 *, 18.5 * | 146.7, 75.3 * |
| C16 Hexadecanoylcarnitine (up) (+) | 10.3, 13.0 * | 5.3, 5.5 * | 13.6, 11.9 * |
| C16:1 Hexadecenoylcarnitine (up) (+) | 200.6, 99.0 * | 31.3 *, 23.2 * | 188.7, 105.7 * |
| C18 Octadecanoylcarnitine (up) (+) | 11.6, 4.5 * | 4.2, 2.1 * | 7.4, 5.1 * |
| C18:2 Linoleoylcarnitine (up) (+) | 15.4, 27.4 * | 8.5, 8.6 * | 33.8, 20.9 * |
| C4 Butyrylcarnitine (up) (+) | 33.5 *, 134.5 * | 10.9 *, 35.1 * | 28.6 *, 182.3 * |
| C5 Isovalerylcarnitine (up) (+) | 362.7 *, 31.7 * | 33.3 *, 17.4 * | 172.3 *, 31.4 * |
| C5DC Glutarylcarnitine (up) (+) | 27.7 *, 91.8 * | 5.8 *, 24.3 * | 25.0 *, 127.0 * |
| C6 Hexanoylcarnitine (up) (+) | 112.9 *, 173.5 * | 28.2 *, 47.1 * | 92.3 *, 200.3 * |
| C8 Octanoylcarnitine (up) (+) | 154.3 *, 32.1 * | 24.6 *, 25.2 * | 114.4 *, 38.4 * |
| Glutaric acid (up) (+) | 1.9 *, 1.1 | 1.0 *, 1.3 | 2.2 *, 1.8 |
| C18:1 Oleoylcarnitine (up) (+) | 16.3, 8.1 | 5.0, 3.9 * | 17.0, 7.0 |
| Homocystinuria N = 3 | |||
| Homocysteine (up) (+) | 2.5 *, 1.8 *, 0.7 * | 2.8 *, 1.2, 0.6 * | 1.5, 1.5, 0.8 * |
| Methionine + Methioninesulfoxide (up) (+) | 7.4 *, 8.5 *, 46.9 * | 8.8 *, 5.2 *, 16.4 * | 8.1 *, 5.6 *, 61.3 * |
| Isovaleric acidemia N = 1 | |||
| C5 Isovalerylcarnitine (up) (+) | 62.6 * | 34.7 * | 39.0 * |
| L−2-Hydroxyglutaric aciduria N = 1 | |||
| Lysine (up) (+) | 5.4 * | 3.5 * | 5.6 * |
| Long-chain−3-hydroxyacyl CoA dehydrogenase deficiency N = 2 | |||
| 3-Hydroxydecanedioic acid (up) (−) | 1.2, 1.4 * | 1.6, 1.6 * | 0.2, −0.1 |
| Adipic acid (1) (up) (−) | 0.8, 0.6 | 0.8, 0.6 | 0.4, −0.0 |
| Sebacic acid (up) (−) | 2.4, 1.2 * | 3.8, 1.2 | 1.4, −0.2 |
| Sebacic acid (up) (+) | 2.9, 1.6 * | 4.0, 1.3 | 1.1, −0.3 |
| Suberic acid (up) (−) | 1.0, 1.0 * | 1.1, 0.8 | 0.2, −0.3 |
| Suberic acid (up) (+) | 1.7, 1.6 * | 1.3, 0.9 | 0.4, −0.1 |
| Lysinuric protein intolerance N = 2 | |||
| Arginine (down) (−) | −1.2 *, −1.6 * | −1.1 *, −1.9 * | −0.8 *, −1.7 * |
| Arginine (down) (+) | −1.2 *, −1.2 * | −1.1 *, −1.9 * | −1.1 *, −1.4 * |
| Glutamine + Glutamic acid/N-Methyl-D-Aspartic acid (up) (−) | 1.3 *, 3.2 * | 0.8, 6.6 * | 0.3, 4.6 * |
| Lysine (down) (+) | −2.1 *, −2.4 * | −2.1 *, −2.8 * | −2.3 *, −1.9 * |
| Ornithine (down) (+) | −0.6 *, −1.6 * | −1.8 *, −2.9 * | −1.2 *, −2.2 * |
| Malonyl-Coa decarboxylase deficiency N = 1 | |||
| Malonic acid (up) (−) | −0.0 | −0.3 | −0.7 * |
| Maple syrup urine disease N = 2 | |||
| (allo)Isoleucine (up) (−) | 3.1 *, 20.2 * | 3.4 *, 9.7 * | 4.3 *, 11.3 * |
| (allo)Isoleucine (up) (+) | 3.6 *, 16.4 * | 3.9 *, 9.9 * | 5.0 *, 11.7 * |
| 2-Keto−3-methylvaleric acid (up) (−) | −1.2 *, 20.5 * | −1.5 *, 14.4 * | −0.8 *, 19.6 * |
| 2-Keto−4-methylvaleric acid (up) (−) | −1.5 *, 0.2 | −2.6 *, 0.2 | −1.5 *, 0.6 |
| Leucine (up) (+) | 3.8 *, 23.8 * | 3.3 *, 13.1 * | 4.4 *, 17.5 * |
| Valine (up) (−) | 0.3, 3.6 | 0.8, 2.8 | 1.7, 3.5 |
| Valine (up) (+) | 2.0 *, 7.5 * | 2.1 *, 5.0 * | 2.0 *, 3.9 * |
| Medium Chain Acyl-CoA Dehydrogenase Deficiency N=5 | |||
| 3-Hydroxydecanedioic acid (up) (−) | 2.0 *, 1.9, 1.9, 0.5, −0.3 | −0.3, −0.3, −0.1, −0.4, −1.8 | 0.7, 0.8, 0.7, −0.1, −0.1 |
| Adipic acid (1) (up) (−) | 2.4 *, 1.3, 1.8, 0.2, −0.3 | 0.2, −0.3, 0.2, −0.7 *, −1.0 | 0.5 *, 0.1, 0.3, −0.5, −0.0 |
| C10:1 Decenoylcarnitine (up) (+) | 31.4 *, 24.3 *, 6.2 *, 19.2 *, 12.8 * | 22.0 *, 18.3 *, 7.6 *, 15.9 *, 8.3 * | 63.3 *, 49.8 *, 13.6 *, 39.4 *, 9.4 * |
| C6 Hexanoylcarnitine (up) (+) | 38.7 *, 32.5 *, 7.7 *, 22.6 *, 5.6 * | 21.4 *, 19.2 *, 7.3 *, 15.0 *, 6.9 * | 78.0 *, 66.1 *, 16.4 *, 46.1 *, 5.6 * |
| C8 Octanoylcarnitine (up) (+) | 58.1 *, 69.7 *, 29.7 *, 38.3 *, 25.2 * | 25.0 *, 27.6 *, 16.5 *, 19.1 *, 12.1 * | 148.7 *, 178.8 *, 76.7 *, 98.2 *, 23.9 * |
| Hexanoic acid/Trans-cyclohexane−1,2-diol (up) (−) | 0.4, 0.1, 0.4, 0.6, −0.4 | −0.1, −0.9, 0.5, 1.3, −0.9 | −0.0, −0.2, 0.0, 0.2, 0.4 |
| Sebacic acid (up) (−) | 0.6, 1.5, 1.3, −0.3, −0.2 | −0.5, −0.3, −0.1, −0.5, −0.9 | 0.0, 0.4, 0.3, −0.2, 0.0 |
| Sebacic acid (up) (+) | 0.1, 0.5, 0.6, −0.5, −0.4 | −0.3, −0.2, 0.1, −0.6, −0.7 | 0.4 *, 0.6, 0.6 *, 0.0, −0.0 |
| Suberic acid (up) (−) | 1.6 *, 1.6, 1.6 *, 0.0, −0.1 | −0.0, −0.2, 0.0, −0.5, −0.7 | 0.3 *, 0.3, 0.3, −0.4, 0.2 |
| Suberic acid (up) (+) | 0.9, 1.0, 0.5, −0.2, −0.5 | 0.1, −0.2, −0.1, −0.6, −0.9 | 0.5, 0.5, 0.2, −0.2, −0.3 |
| Methylmalonyl-CoA mutase deficiency N = 1 | |||
| C3 Propionylcarnitine (up) (+) | 85.0 * | 39.9 * | 234.9 * |
| Organic cation transporter 2 deficiency N = 1 | |||
| C0 L-Carnitine (down) (+) | −2.3 * | −1.3 * | −0.7 |
| Ornithine aminotransferase N = 1 | |||
| Guanidinoacetic acid (down) (+) | −2.2 * | −2.2 * | −1.7 * |
| Ornithine (up) (+) | 33.1 * | 11.7 * | 37.4 * |
| Ornithine transcarbamylase deficiency N = 2 | |||
| Citrulline (down) (−) | 2.9 *, 1.0 * | 1.4 *, 0.6 | 2.0 *, 1.1 * |
| Citrulline (down) (+) | 1.4 *, 2.5 * | 0.6 *, 1.1 * | 1.2 *, 1.5 * |
| Glutamine + Glutamic acid/N-Methyl-D-Aspartic acid (up) (−) | 1.0 *, 1.3 * | 0.6, 0.6 | 0.3, 1.5 * |
| Uridine (up) (−) | 7.7 *, 4.0 * | 4.4 *, 5.0 * | 6.5 *, 5.4 * |
| Phenylketonuria N = 4 | |||
| N-Acetylphenylalanine (up) (−) | 116.5 *, 18.6, 73.9 *, 8.5 * | 25.2 *, 11.9 *, 34.6 *, 7.3 * | 45.7 *, 14.7, 56.3 *, 6.5 * |
| Phenylacetic acid (up) (+) | −0.3, −0.1, 1.6, 0.6 | −0.4, −0.0, 1.6, 0.6 | −0.3, −0.1, 0.1, 1.2 |
| Phenylalanine (up) (−) | 87.7 *, 38.5 *, 161.6 *, 14.6 * | 34.3 *, 23.4 *, 43.4 *, 9.5 * | 94.2 *, 51.6 *, 121.7 *, 26.1 * |
| Phenylalanine (up) (+) | 42.6 *, 24.5 *, 80.8 *, 30.3 * | 21.1 *, 16.7 *, 29.9 *, 9.2 * | 55.5 *, 38.3 *, 68.9 *, 22.9 * |
| alpha-N-Phenylacetylglutamine (up) (−) | 5.2 *, 1.3 *, 4.7 *, 0.8 | 2.2 *, 1.6 *, 3.2 *, 0.3 | 3.7 *, 2.4 *, 5.2 *, −1.0 * |
| alpha-N-Phenylacetylglutamine (up) (+) | 6.1 *, 1.4, 4.3 *, 0.4 | 2.8 *, 1.9 *, 3.1 *, 0.3 | 4.9 *, 3.1, 4.7 *, −0.7 * |
| Propionic acidemia N = 2 | |||
| C3 Propionylcarnitine (up) (+) | 124.3 *, 199.2 * | 47.8 *, 37.1 * | 148.5 *, 200.1 * |
| Thymidine phosphorylase deficiency N = 1 | |||
| Thymidine (up) (−) | 44.3 * | 103.5 * | 35.3 * |
| Tyrosinemia I N = 2 | |||
| 4-Hydroxyphenyllactic acid (up) (−) | 337.5 *, 1046.6 * | 11.4 *, 16.1 * | 403.6 *, 582.8 * |
| Tyrosine (up) (+) | 16.6 *, 43.1 * | 9.8 *, 23.6 * | 25.2 *, 40.4 * |
| Very Long Chain Acyl-CoA Dehydrogenase Deficiency N = 1 | |||
| C14:1 Tetradecenoylcarnitine (up) (+) | 211.0 * | 23.8 * | 213.8 * |
| C1 6 Hexadecanoylcarnitine (up) (+) | 7.0 * | 2.3 * | 7.3 * |
| C18 Octadecanoylcarnitine (up) (+) | 3.3 * | 1.2 * | 3.9 * |
| C18:1 Oleoylcarnitine (up) (+) | 5.8 * | 2.0 * | 9.5 * |
| Carnitine palmitoyltransferase II N = 2 | |||
| Adipic acid (1) (up) (−) | −0.1, 0.1 | −0.0, −0.1 | 0.7, −0.0 |
| C0 L-Carnitine (down) (+) | −0.1, −0.7 | −0.1, −1.0 * | 1.5 *, −0.4 |
| C16 Hexadecanoylcarnitine (up) (+) | 8.5 *, 19.3 * | 4.1 *, 6.8 * | 8.0 *, 30.9 * |
| C18 Octadecanoylcarnitine (up) (+) | 13.8 *, 53.5 * | 5.0 *, 8.2 * | 14.4 *, 45.3 * |
| Sebacic acid (up) (−) | −0.1, −0.3 | −0.0, −0.4 | 0.7, −0.3 |
| Sebacic acid (up) (+) | 0.2, 0.1 | −0.1, −0.2 | 0.7, −0.2 |
| Suberic acid (up) (−) | 0.1, −0.2 | 0.3, −0.4 | 1.0, −0.2 |
| Suberic acid (up) (+) | 0.2, −0.5 | 0.0, −0.5 | 0.5, −0.2 |
| C18:1 Oleoylcarnitine (up) (+) | 6.5 *, 27.7 * | 3.5 *, 5.6 * | 5.7 *, 33.8 * |
Appendix E. Lost Biomarkers Due to Merging of Datasets
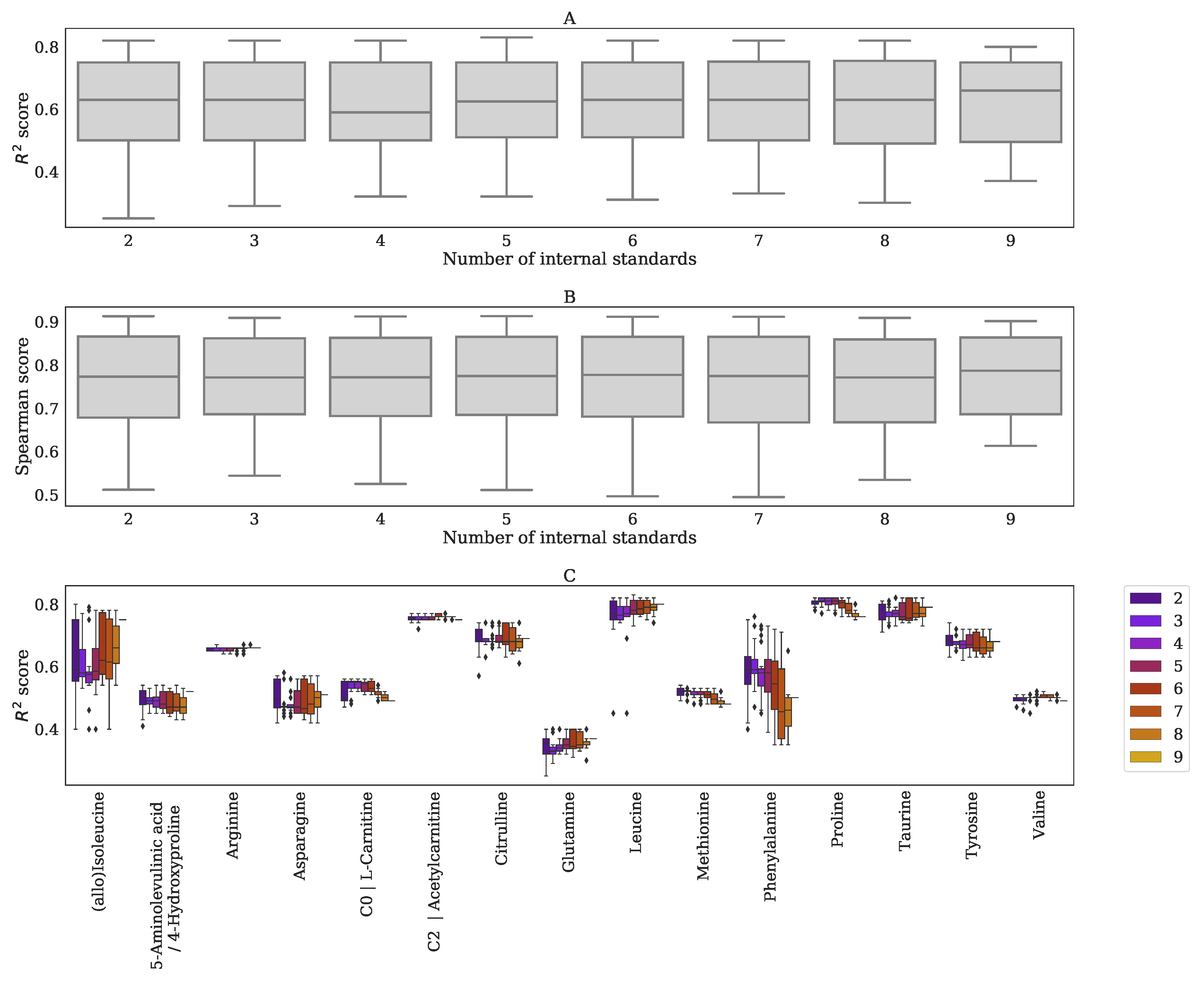
Appendix F. Performance BC-Metchalizer for a Different Number of Internal Standards

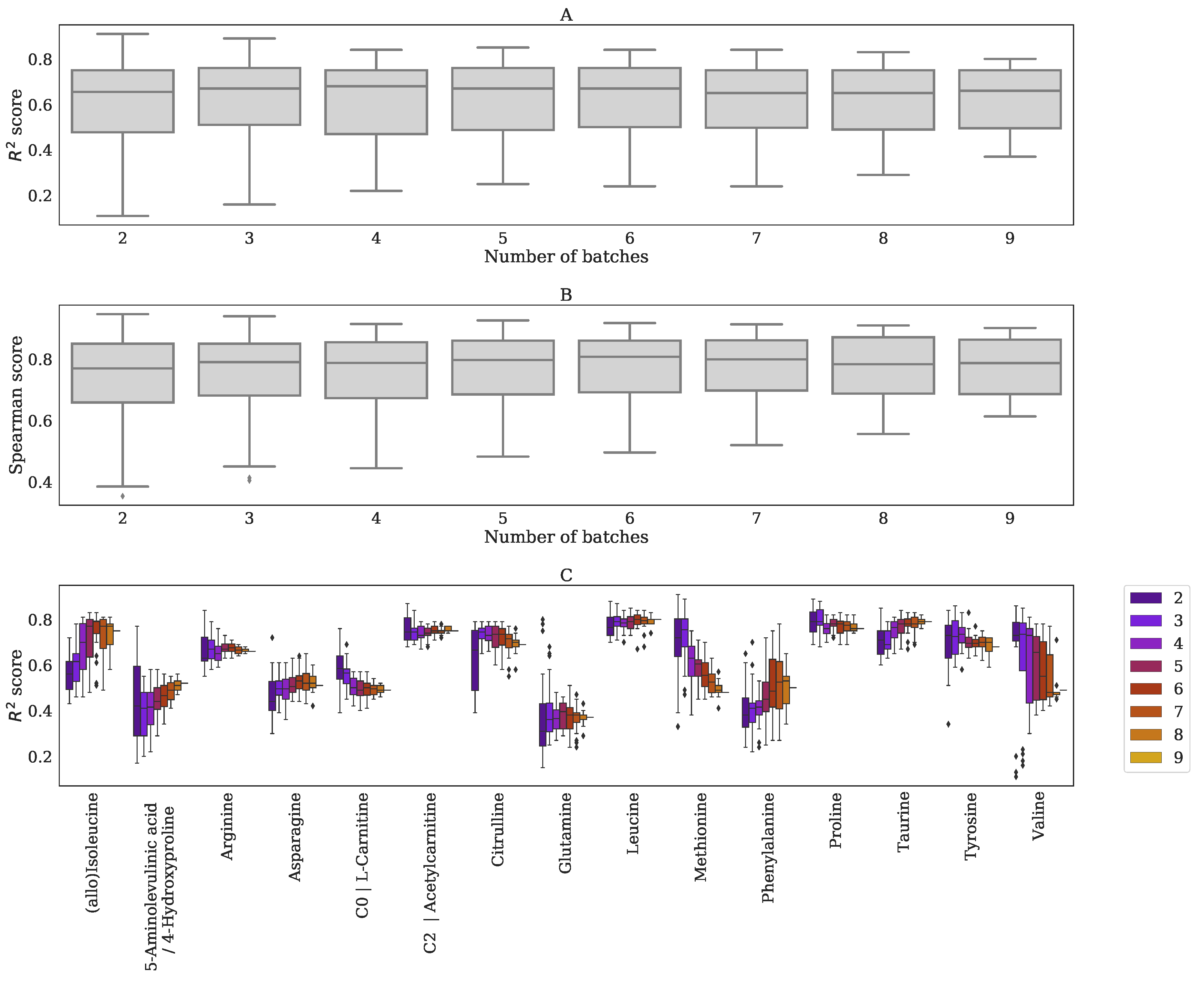
Appendix G. Performance BC-Metchalizer for a Different Number of Batches

Appendix H. Age and Sex Similarity of Reference Samples with Patients for Different Z-Score Methods

Appendix I. The Influence of Log-Transformation on Z-Scores and p-Values

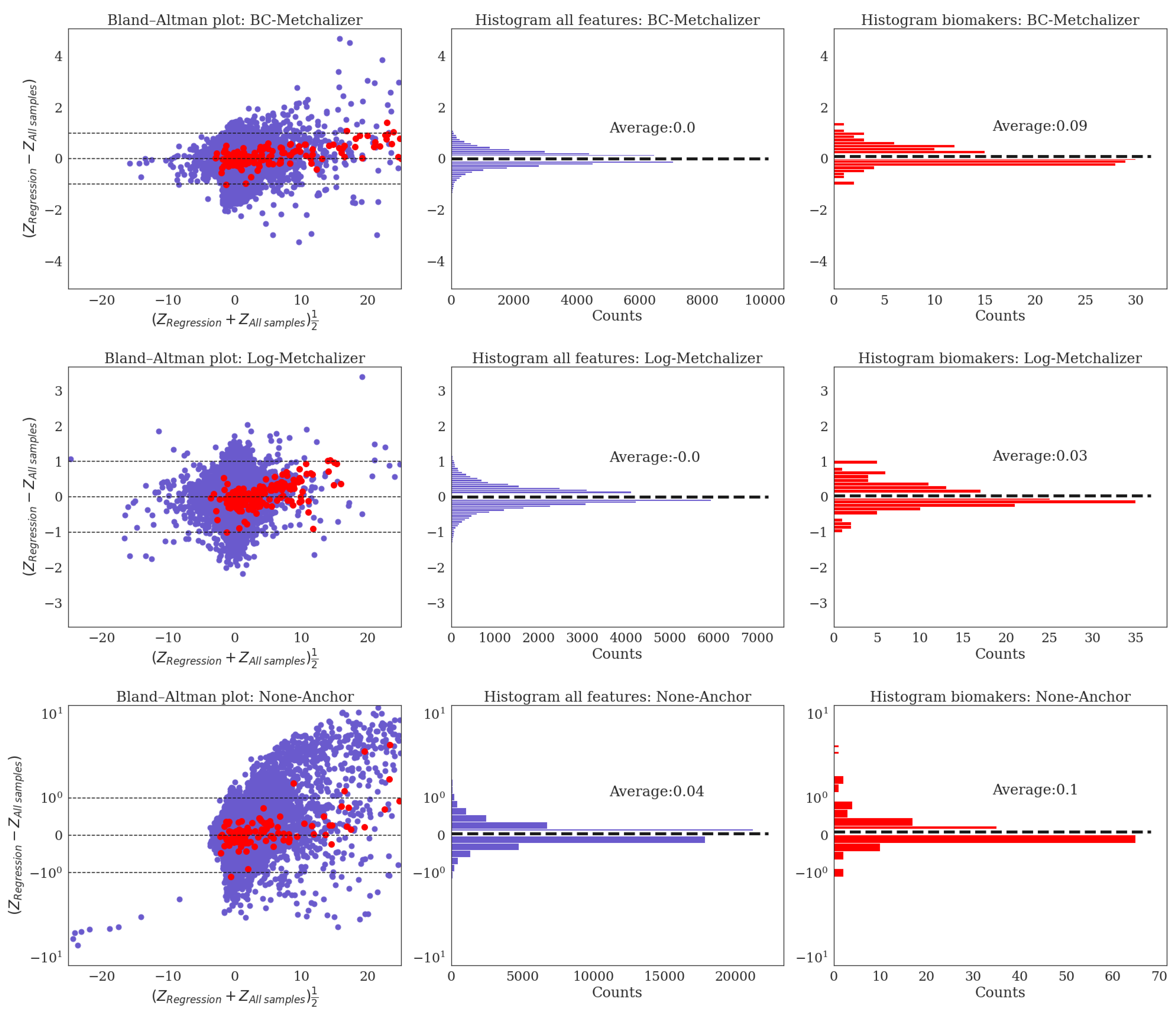
Appendix J. Bland–Altman Analysis for Z-Scores Obtained Using All Samples Versus Regression

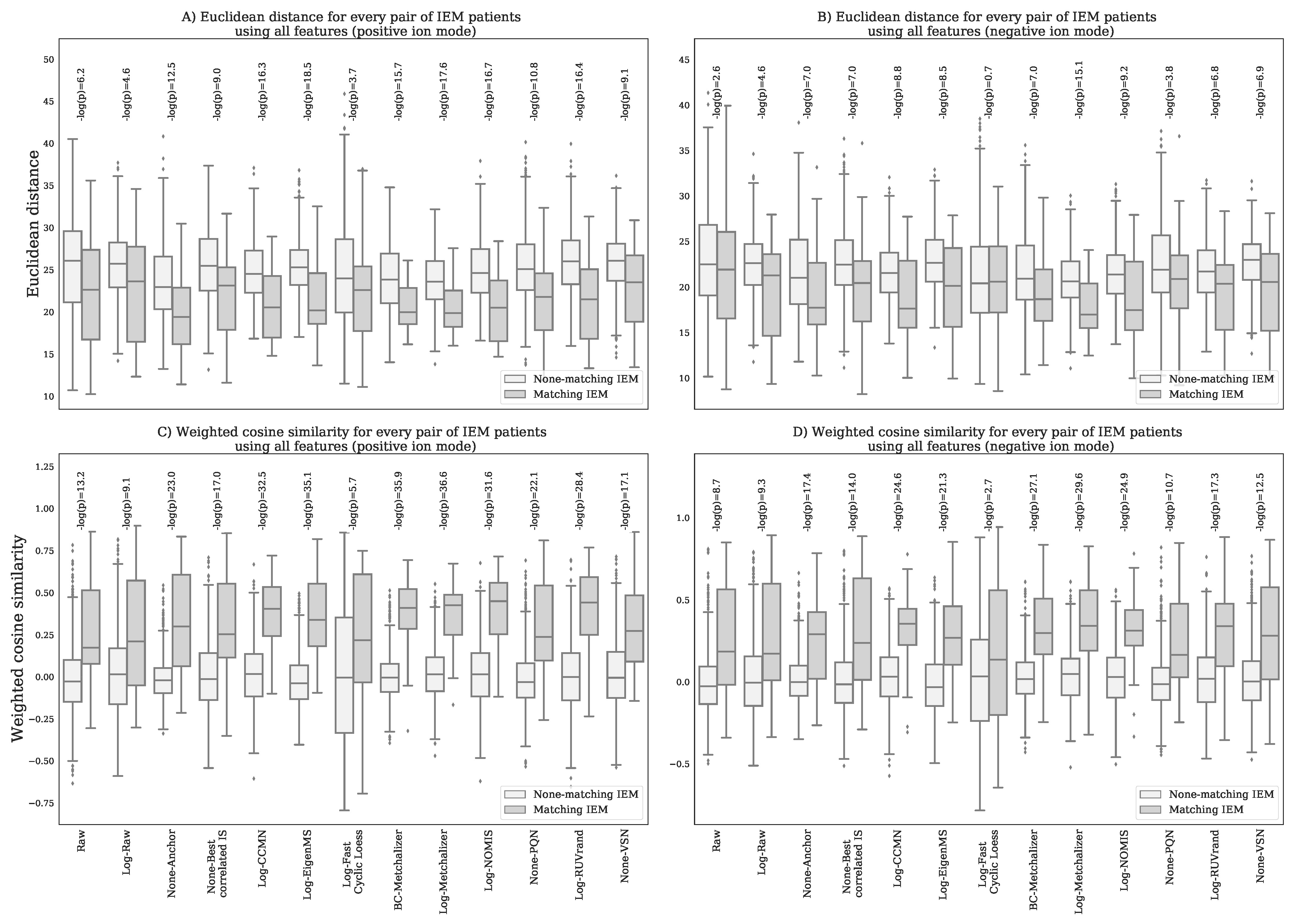
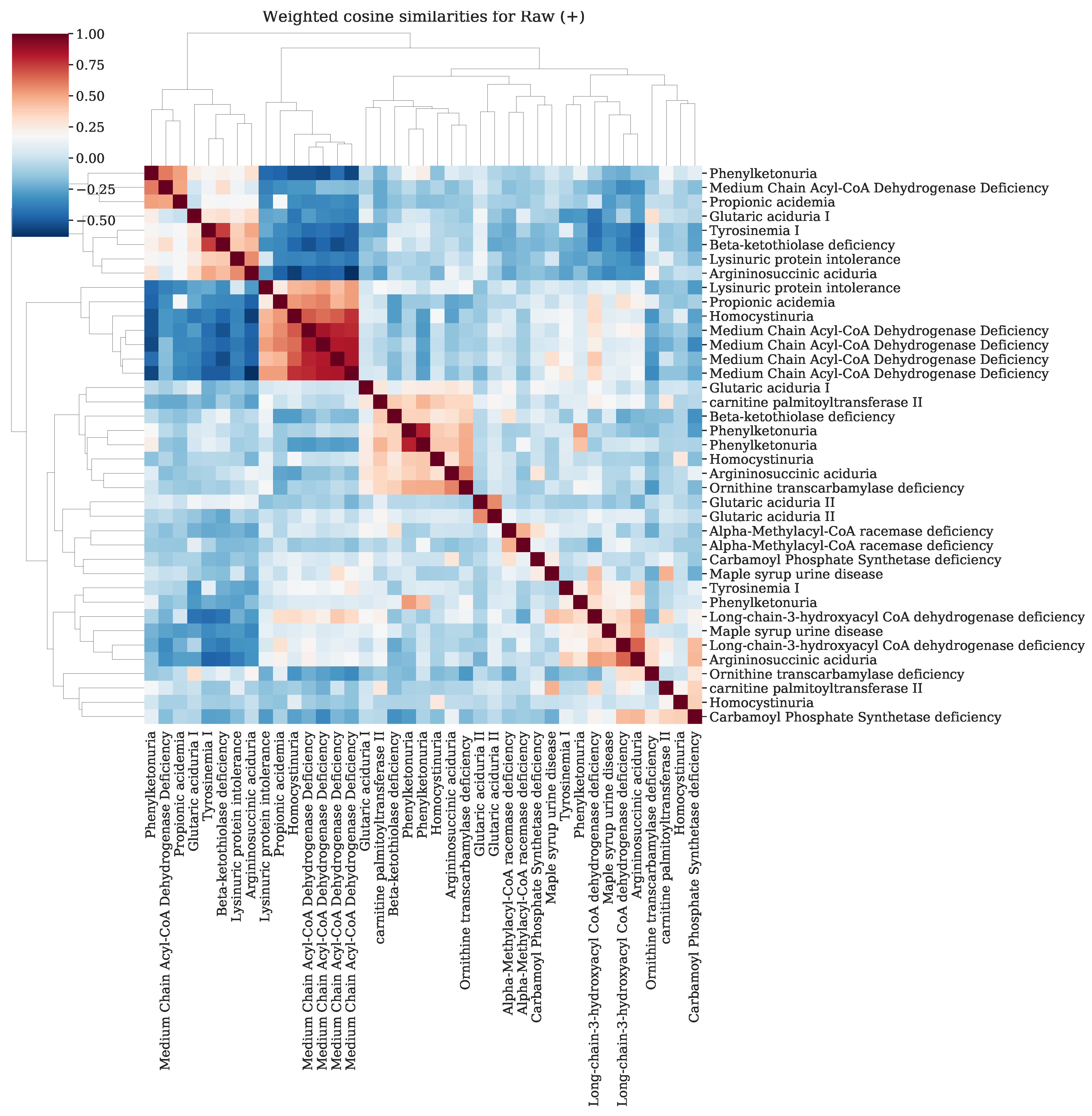
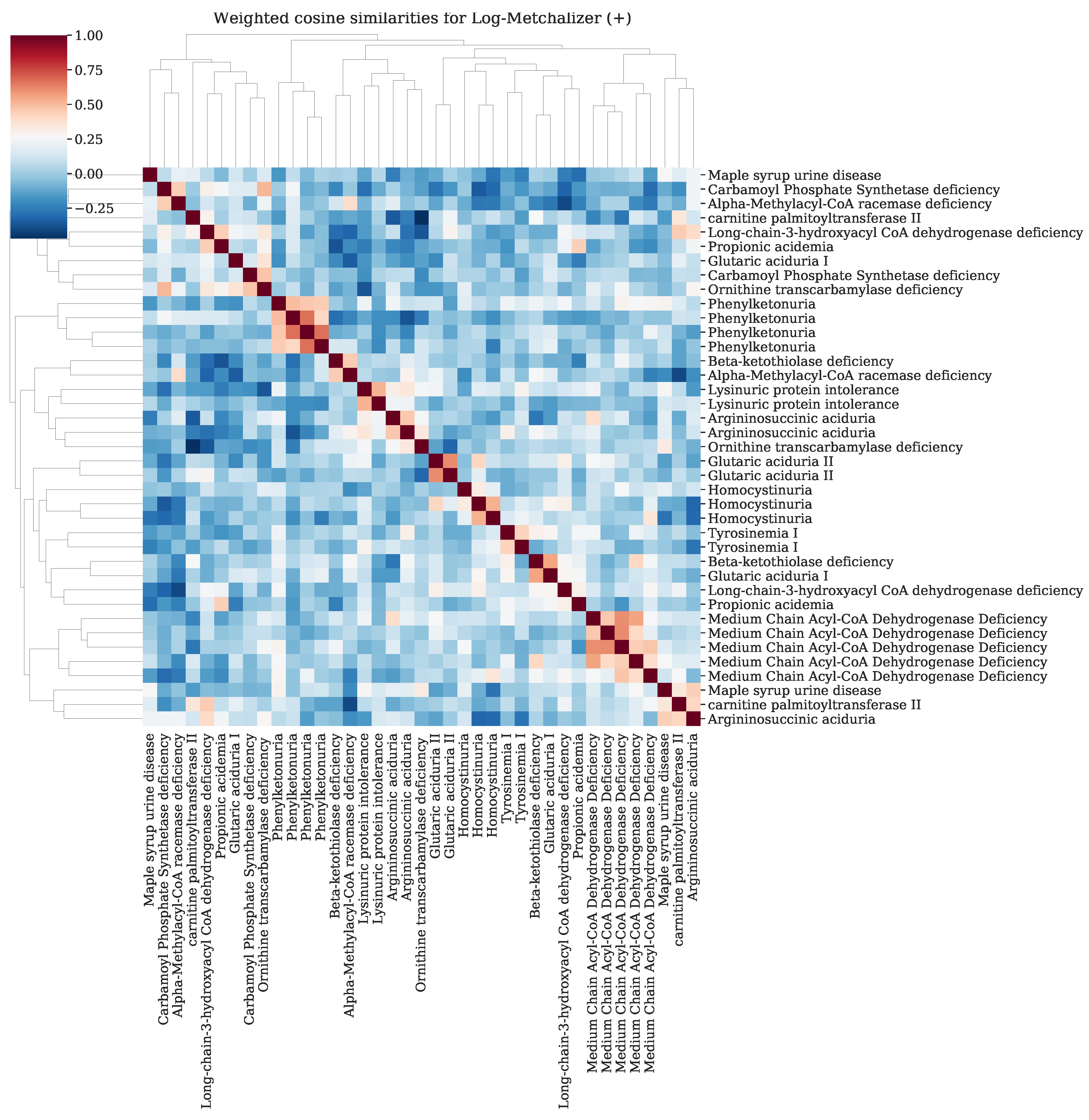
Appendix K. Resemblance of Patients Sharing the Same IEM



References
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Körver-Keularts, I.M.L.W.; Wang, P.; Waterval, H.W.A.H.; Kluijtmans, L.A.J.; Wevers, R.A.; Langhans, C.D.; Scott, C.; Habets, D.D.J.; Bierau, J. Fast and accurate quantitative organic acid analysis with LC-QTOF/MS facilitates screening of patients for inborn errors of metabolism. J. Inherit. Metab. Dis. 2018, 41, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Willemsen, M.; Van der Ham, M.; Gerrits, J.; Pras-Raves, M.L.; Prinsen, H.C.M.T.; Van Hasselt, P.M.; De Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.; et al. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Bonte, R.; Bongaerts, M.; Demirdas, S.; Langendonk, J.G.; Huidekoper, H.H.; Williams, M.; Onkenhout, W.; Jacobs, E.H.; Blom, H.J.; Ruijter, G.J.G. Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform. Metabolites 2019, 9, 289. [Google Scholar] [CrossRef]
- Glinton, K.E.; Levy, H.L.; Kennedy, A.D.; Pappan, K.L.; Elsea, S.H. Untargeted metabolomics identifies unique though benign biochemical changes in patients with pathogenic variants in UROC1. Mol. Genet. Metab. Rep. 2019, 18, 14–18. [Google Scholar] [CrossRef]
- Chaleckis, R.; Murakami, I.; Takada, J.; Kondoh, H.; Yanagida, M. Individual variability in human blood metabolites identifies age-related differences. Proc. Natl. Acad. Sci. USA 2016, 113, 4252–4259. [Google Scholar] [CrossRef]
- Rist, M.J.; Roth, A.; Frommherz, L.; Weinert, C.H.; Krüger, R.; Merz, B.; Bunzel, D.; Mack, C.; Egert, B.; Bub, A.; et al. Metabolite patterns predicting sex and age in participants of the Karlsruhe Metabolomics and Nutrition (KarMeN) study. PLoS ONE 2017, 12, e0183228. [Google Scholar] [CrossRef]
- Yu, Z.; Zhai, G.; Singmann, P.; He, Y.; Xu, T.; Prehn, C.; Römisch-Margl, W.; Lattka, E.; Gieger, C.; Soranzo, N.; et al. Human serum metabolic profiles are age dependent. Aging Cell 2012, 11, 960–967. [Google Scholar] [CrossRef]
- Lawton, K.A.; Berger, A.; Mitchell, M.; Milgram, K.E.; Evans, A.M.; Guo, L.; Hanson, R.W.; Kalhan, S.C.; Ryals, J.A.; Milburn, M.V. Analysis of the adult human plasma metabolome. Pharmacogenomics 2008, 9, 383–397. [Google Scholar] [CrossRef]
- Veselkov, K.A.; Vingara, L.K.; Masson, P.; Robinette, S.L.; Want, E.; Li, J.V.; Barton, R.H.; Boursier-Neyret, C.; Walther, B.; Ebbels, T.M.; et al. Optimized Preprocessing of Ultra-Performance Liquid Chromatography/Mass Spectrometry Urinary Metabolic Profiles for Improved Information Recovery. Anal. Chem. 2011, 83, 5864–5872. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tang, J.; Yang, Q.; Li, S.; Cui, X.; Li, Y.; Chen, Y.; Xue, W.; Li, X.; Zhu, F. NOREVA: Normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017, 45, W162–W170. [Google Scholar] [CrossRef] [PubMed]
- Välikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Briefings Bioinform. 2016, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vreken, P.; van Lint, A.E.M.; Bootsma, A.H.; Overmars, H.; Wanders, R.J.A.; van Gennip, A.H. Rapid Diagnosis of Organic Acidemias and Fatty-acid Oxidation Defects by Quantitative Electrospray Tandem-MS Acyl-Carnitine Analysis in Plasma. In Current Views of Fatty Acid Oxidation and Ketogenesis; Springer US: New York, NY, USA, 2002; pp. 327–337. [Google Scholar] [CrossRef]
- Redestig, H.; Fukushima, A.; Stenlund, H.; Moritz, T.; Arita, M.; Saito, K.; Kusano, M. Compensation for Systematic Cross-Contribution Improves Normalization of Mass Spectrometry Based Metabolomics Data. Anal. Chem. 2009, 81, 7974–7980. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Nikolic, S.B.; Wilson, R.; Sharman, J.E.; Edwards, L.M. Metabolomics Data Normalization with EigenMS. PLoS ONE 2015, 9, e116221. [Google Scholar] [CrossRef]
- Ballman, K.V.; Grill, D.E.; Oberg, A.L.; Therneau, T.M. Faster cyclic loess: Normalizing RNA arrays via linear models. Bioinformatics 2004, 20, 2778–2786. [Google Scholar] [CrossRef]
- Sysi-Aho, M.; Katajamaa, M.; Yetukuri, L.; Orešič, M. Normalization method for metabolomics data using optimal selection of multiple internal standards. BMC Bioinform. 2007, 8, 93. [Google Scholar] [CrossRef]
- Filzmoser, P.; Walczak, B. What can go wrong at the data normalization step for identification of biomarkers? J. Chromatogr. A 2014, 1362, 194–205. [Google Scholar] [CrossRef]
- Livera, A.M.D.; Sysi-Aho, M.; Jacob, L.; Gagnon-Bartsch, J.A.; Castillo, S.; Simpson, J.A.; Speed, T.P. Statistical Methods for Handling Unwanted Variation in Metabolomics Data. Anal. Chem. 2015, 87, 3606–3615. [Google Scholar] [CrossRef]
- Huber, W.; von Heydebreck, A.; Sultmann, H.; Poustka, A.; Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]






| Coefficient | Ion Mode | None-Anchor | BC-Metchalizer | Log-Metchalizer |
|---|---|---|---|---|
| − | 98.4 | 100.0 | 100.0 | |
| + | 100.0 | 100.0 | 100.0 | |
| − | 22.9 | 36.9 | 31.8 | |
| + | 6.5 | 12.5 | 13.2 | |
| − | 4.5 | 17.8 | 16.9 | |
| + | 0.5 | 4.4 | 5.1 | |
| − | 1.6 | 8.3 | 8.3 | |
| + | 0.2 | 0.2 | 0.5 | |
| − | 0.0 | 0.0 | 0.0 | |
| + | 0.0 | 0.0 | 0.0 | |
| − | 0.0 | 0.0 | 0.0 | |
| + | 0.5 | 0.7 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bongaerts, M.; Bonte, R.; Demirdas, S.; Jacobs, E.H.; Oussoren, E.; van der Ploeg, A.T.; Wagenmakers, M.A.E.M.; Hofstra, R.M.W.; Blom, H.J.; Reinders, M.J.T.; et al. Using Out-of-Batch Reference Populations to Improve Untargeted Metabolomics for Screening Inborn Errors of Metabolism. Metabolites 2021, 11, 8. https://doi.org/10.3390/metabo11010008
Bongaerts M, Bonte R, Demirdas S, Jacobs EH, Oussoren E, van der Ploeg AT, Wagenmakers MAEM, Hofstra RMW, Blom HJ, Reinders MJT, et al. Using Out-of-Batch Reference Populations to Improve Untargeted Metabolomics for Screening Inborn Errors of Metabolism. Metabolites. 2021; 11(1):8. https://doi.org/10.3390/metabo11010008
Chicago/Turabian StyleBongaerts, Michiel, Ramon Bonte, Serwet Demirdas, Edwin H. Jacobs, Esmee Oussoren, Ans T. van der Ploeg, Margreet A. E. M. Wagenmakers, Robert M. W. Hofstra, Henk J. Blom, Marcel J. T. Reinders, and et al. 2021. "Using Out-of-Batch Reference Populations to Improve Untargeted Metabolomics for Screening Inborn Errors of Metabolism" Metabolites 11, no. 1: 8. https://doi.org/10.3390/metabo11010008
APA StyleBongaerts, M., Bonte, R., Demirdas, S., Jacobs, E. H., Oussoren, E., van der Ploeg, A. T., Wagenmakers, M. A. E. M., Hofstra, R. M. W., Blom, H. J., Reinders, M. J. T., & Ruijter, G. J. G. (2021). Using Out-of-Batch Reference Populations to Improve Untargeted Metabolomics for Screening Inborn Errors of Metabolism. Metabolites, 11(1), 8. https://doi.org/10.3390/metabo11010008