Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage


Abstract
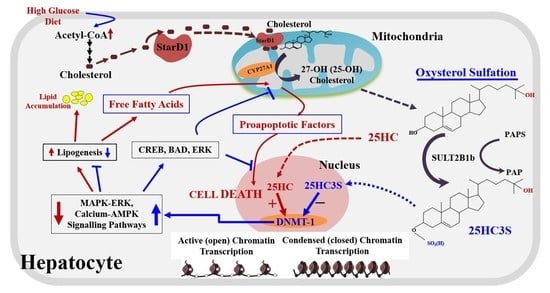
{kind=link}
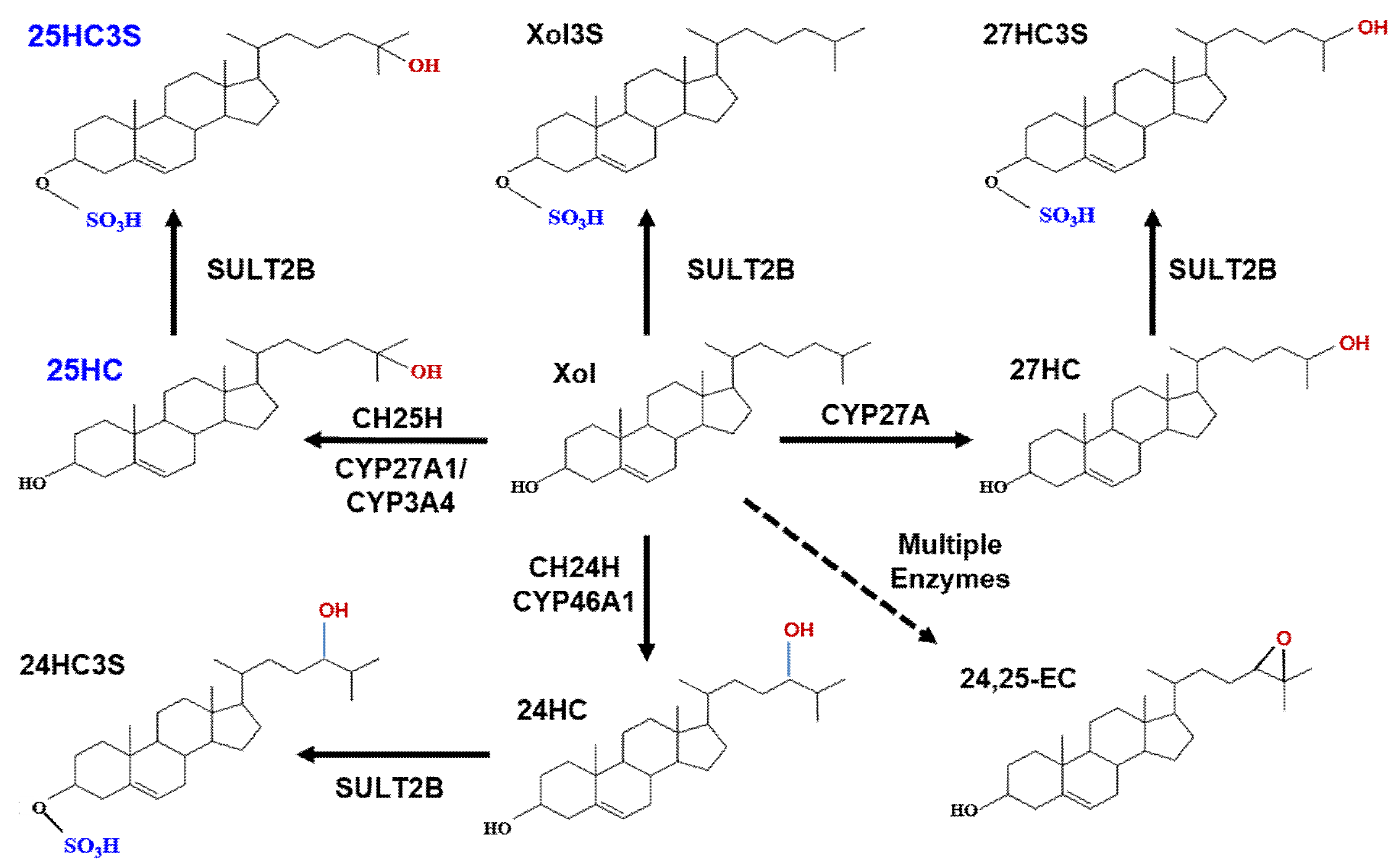{kind=link}
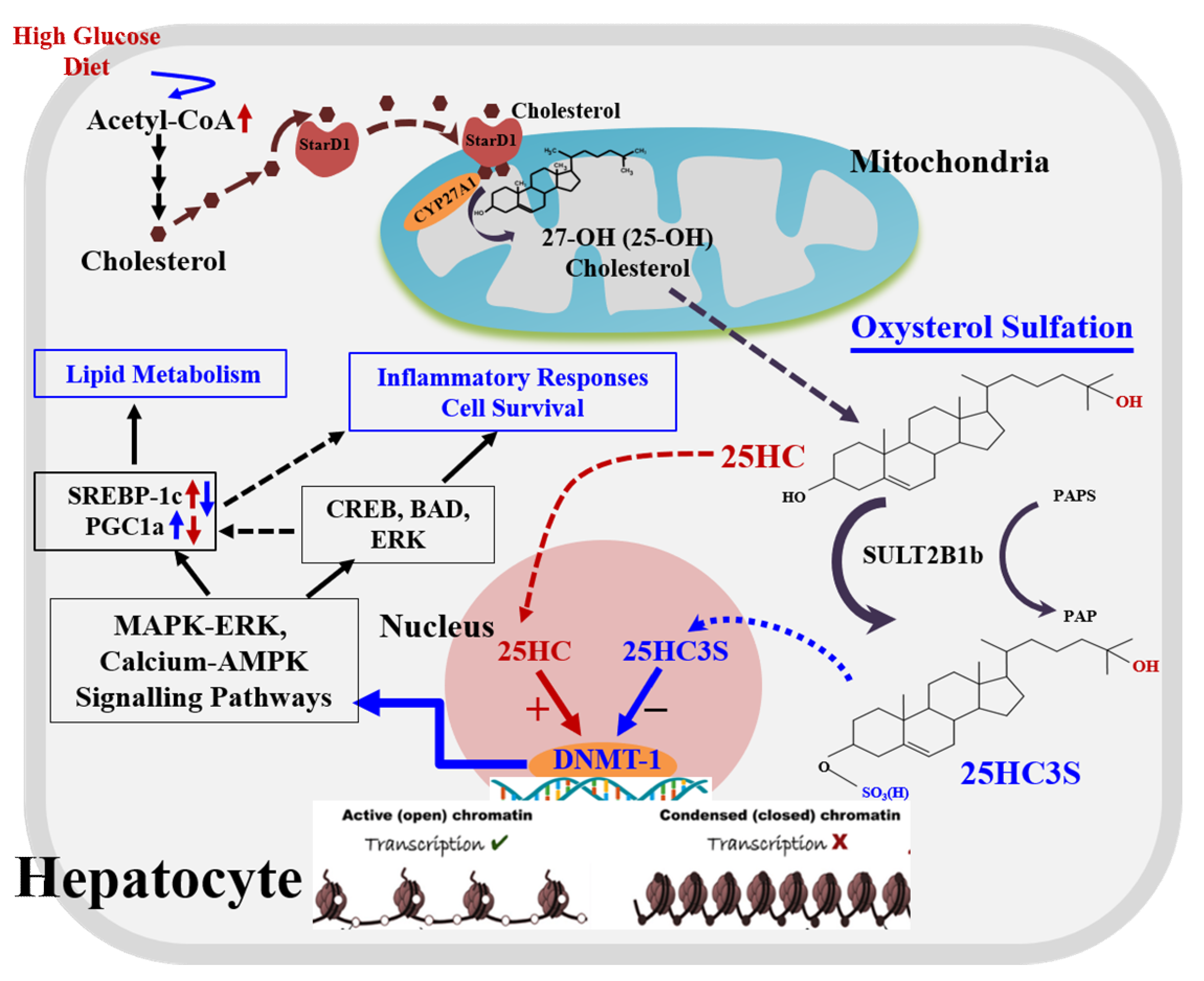{kind=link}
{kind=link}
1. Introduction
2. 25-Hydroxycholesterol Induces Lipogenesis and Cell Apoptosis
3. Discovery of Oxysterol Sulfates and Exploration of Their Function
4. 25HC3S/25HCDS Suppresses Cholesterol and Triglyceride Biosynthesis in Hepatocytes
5. 25HC3S Increases Nuclear PPARγ Levels and Suppresses Inflammatory Responses via the PPARγ/IκBα Signaling Pathway
6. Biochemical Mechanism: 25HC and 25HC3S Are Paired Epigenetic Regulators
7. In Vivo Studies: 25HC3S/25HCDS Has Potential for Clinical Use
8. 25HC3S Decreases Lipid Accumulation, Suppresses Inflammatory Responses, and Decreases Liver Fibrosis in Mouse NAFLD Models
9. 25HC3S and Oxysterol Sulfation Suppress Hepatocyte Apoptosis and Promotes Regeneration in Mouse Hepatectomy Model
10. 25HC3S/25HCDS Alleviate LPS—Induced Acute Organ Injury in Mouse Model
11. Clinical Development: 25HC3S to Serve as a Treatment in Chronic Liver Diseases
12. Phase 1 Study: Safety and Pharmacokinetics of DUR-928 in Healthy Human Volunteers
13. Phase 1b Study: DUR-928 in Liver Function-Impaired (NASH) Patients
14. Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| 24HC | 24-Hydroxycholesterol |
| 24HC3S | 24-Hydroxycholesterol 3-sulfate |
| 25HC | 25-Hydroxycholesterol |
| 25HC | 25-Hydroxycholesterol |
| 25HC3S | 25-Hydroxyvholesterol 3-sulfate |
| 25HCDS | 5-Cholesten-3β, 25-diol-disulfate |
| 27HC | 27-Hydroxycholesterol |
| 27HC3S | 27-Hydroxycholesterol 3-sulfate |
| 7-alpha, 25-DC | 7-Alpha, 25-dihydroxycholesterol |
| ABCA1 | ATP-binding cassette transporter A1 |
| ACC | Acetyl-CoA carboxylase |
| AOI | Acute organ injury |
| Apaf1 | Apoptotic peptidase activating factor 1 |
| C24HL | Cholesterol 24-hydroxidase |
| CDC25b | Cell Division Cycle 25B |
| CH25L | Cholesterol 25-hydroxylase |
| CK-18 | Cytokeratin-18 |
| CpG | 5’—cytosine—phosphate—guanine—3’ |
| CYP27A1 | Cytochrome P450 Family 27 Subfamily A Member 1 |
| CYP3A4 - | Cytochrome P450 3A4 |
| CYP46A1 | Cytochrome P450 46A1 |
| CYP7A1 | Cholesterol 7α-hydroxylase |
| DNMT | DNA methyltransferases |
| DUR-928 | 25HC3S |
| FAS | Fatty acid synthase |
| FFA | Free fatty acids |
| FoxM1b | Forkhead Box (Fox) m1b |
| GPAM | Glycerol-3-phosphate acyltransferase 1, mitochondrial |
| HFD | High fat diet |
| HMGR | 3-Hydroxy-3-methylglutaryl coenzyme-A reductase |
| HPLC | High-performance liquid chromatography |
| hsCRP | High sensitivity C-reactive protein |
| IL1β | Interleukin 1 beta |
| IκBα | Inhibitor kappa B-alpha |
| LDLR | Low density lipoprotein receptor |
| LXR | Liver x receptor |
| MS/MS | mass/mass spectrometry |
| NAFLD | Nonalcoholic fatty liver diseases |
| NASH | Nonalcoholic steatohepatitis |
| NF-Κb | Nuclear factor-kappa B |
| PCNA | Proliferating cell nuclear antigen |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| SREBP | Sterol regulatory element binding protein |
| STS | Steroid sulfatase |
| SULT2A1 | Sulfotransferase 2A1 |
| SULT2B1b | Sulfotransferase 2B1b |
| TNFα | Tumor Necrosis Factor α |
| Xol3S | Cholesterol 3-sulfate |
References
- Brown, A.J.; Jessup, W. Oxysterols and atherosclerosis. Atherosclerosis 1999, 142, 1–28. [Google Scholar] [CrossRef]
- Björkhem, I.; Meaney, S.; Diczfalusy, U. Oxysterols in human circulation: Which role do they have? Curr. Opin. Lipidol. 2002, 13, 247–253. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Wang, Y. Oxysterol research: A brief review. Biochem. Soc. Trans. 2019, 47, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Wollam, J.; Antebi, A. Sterol regulation of metabolism, homeostasis, and development. Annu. Rev. Biochem. 2011, 80, 885–916. [Google Scholar] [CrossRef] [PubMed]
- Schroepfer, G.J., Jr. Oxysterols: Modulators of cholesterol metabolism and other processes. Physiol. Rev. 2000, 80, 361–554. [Google Scholar] [CrossRef]
- Oguro, H. The Roles of Cholesterol and Its Metabolites in Normal and Malignant Hematopoiesis. Front. Endocrinol. 2019, 10, 204. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Wang, Y. An update on oxysterol biochemistry: New discoveries in lipidomics. Biochem. Biophys. Res. Commun. 2018, 504, 617–622. [Google Scholar] [CrossRef]
- Lund, E.G.; Kerr, T.A.; Sakai, J.; Li, W.P.; Russell, D.W. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J. Biol. Chem. 1998, 273, 34316–34327. [Google Scholar] [CrossRef]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.M.; Liang, G.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef]
- Russell, D.W. Oxysterol biosynthetic enzymes. Biochim. Biophys. Acta 2000, 1529, 126–135. [Google Scholar] [CrossRef]
- Tabas, I. Cholesterol in health and disease. J. Clin. Investig. 2002, 110, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Quinn, C.M.; Brown, A.J. Synthesis of the oxysterol, 24(S), 25-epoxycholesterol, parallels cholesterol production and may protect against cellular accumulation of newly-synthesized cholesterol. Lipids Health Dis. 2007, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Björkhem, I.; Diczfalusy, U. Oxysterols: Friends, foes, or just fellow passengers? Arter. Thromb. Vasc. Biol. 2002, 22, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Mutemberezi, V.; Muccioli, G.G. Oxysterols in Metabolic Syndrome: From Bystander Molecules to Bioactive Lipids. Trends Mol. Med. 2016, 22, 594–614. [Google Scholar] [CrossRef] [PubMed]
- Vurusaner, B.; Leonarduzzi, G.; Gamba, P.; Poli, G.; Basaga, H. Oxysterols and mechanisms of survival signaling. Mol. Asp. Med. 2016, 49, 8–22. [Google Scholar] [CrossRef]
- Poli, G.; Biasi, F.; Leonarduzzi, G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013, 1, 125–130. [Google Scholar] [CrossRef]
- Hughes, T.M.; Rosano, C.; Evans, R.W.; Kuller, L.H. Brain cholesterol metabolism, oxysterols, and dementia. J. Alzheimers Dis. 2013, 33, 891–911. [Google Scholar] [CrossRef]
- Bai, Q.; Zhang, X.; Xu, L.; Kakiyama, G.; Heuman, D.; Sanyal, A.; Pandak, W.M.; Yin, L.; Xie, W.; Ren, S. Oxysterol sulfation by cytosolic sulfotransferase suppresses liver X receptor/sterol regulatory element binding protein-1c signaling pathway and reduces serum and hepatic lipids in mouse models of nonalcoholic fatty liver disease. Metab. Clin. Exp. 2012, 61, 836–845. [Google Scholar] [CrossRef]
- Li, X.; Pandak, W.M.; Erickson, S.K.; Ma, Y.; Yin, L.; Hylemon, P.; Ren, S. Biosynthesis of the regulatory oxysterol, 5-cholesten-3beta,25-diol 3-sulfate, in hepatocytes. J. Lipid Res. 2007, 48, 2587–2596. [Google Scholar] [CrossRef]
- Sanchez, L.D.; Pontini, L.; Marinozzi, M. Cholesterol and oxysterol sulfates: Pathophysiological roles and analytical challenges. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Cook, I.T.; Duniec-Dmuchowski, Z.; Kocarek, T.A.; Runge-Morris, M.; Falany, C.N. 24-hydroxycholesterol sulfation by human cytosolic sulfotransferases: Formation of monosulfates and disulfates, molecular modeling, sulfatase sensitivity, and inhibition of liver x receptor activation. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Runge-Morris, M.; Kocarek, T.A.; Falany, C.N. Regulation of the cytosolic sulfotransferases by nuclear receptors. Drug Metab. Rev. 2013, 45, 15–33. [Google Scholar] [CrossRef]
- Ren, S.; Hylemon, P.; Zhang, Z.P.; Rodriguez-Agudo, D.; Marques, D.; Li, X.; Zhou, H.; Gil, G.; Pandak, W.M. Identification of a novel sulfonated oxysterol, 5-cholesten-3beta,25-diol 3-sulfonate, in hepatocyte nuclei and mitochondria. J. Lipid Res. 2006, 47, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Kim, J.K.; Kakiyama, G.; Rodriguez-Agudo, D.; Pandak, W.M.; Min, H.K.; Ning, Y. Identification of novel regulatory cholesterol metabolite, 5-cholesten, 3beta,25-diol, disulfate. PLoS ONE 2014, 9, e103621. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Miyazaki, T.; Ikegami, T.; Iwamoto, J.; Maeda, T.; Hirayama, T.; Saito, Y.; Teramoto, T.; Matsuzaki, Y. Cholesterol 25-hydroxylation activity of CYP3A. J. Lipid Res. 2011, 52, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, S.; Abreu de Oliveira, W.A.; Yang, S.; Yutuc, E.; Saeed, A.; Abdel-Khalik, J.; Ullgren, A.; Cedazo-Minguez, A.; Björkhem, I.; Wang, Y.; et al. 24(S),25-Epoxycholesterol and cholesterol 24S-hydroxylase (CYP46A1) overexpression promote midbrain dopaminergic neurogenesis in vivo. J. Biol. Chem. 2019, 294, 4169–4176. [Google Scholar] [CrossRef] [PubMed]
- Emgard, J.; Kammoun, H.; Garcia-Cassani, B.; Chesne, J.; Parigi, S.M.; Jacob, J.M.; Cheng, H.W.; Evren, E.; Das, S.; Czarnewski, P.; et al. Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity 2018, 48, 120–132.e128. [Google Scholar] [CrossRef]
- Chen, J.J.; Lukyanenko, Y.; Hutson, J.C. 25-hydroxycholesterol is produced by testicular macrophages during the early postnatal period and influences differentiation of Leydig cells in vitro. Biol. Reprod. 2002, 66, 1336–1341. [Google Scholar] [CrossRef]
- Janowski, B.; Willy, P.; Devi, T.; Falck, J.; Mangelsdorf, D. An Oxysterol Signaling Pathway Mediated by the Nuclear Receptor Lxr-Alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Zhu, R.; Ou, Z.; Ruan, X.; Gong, J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review). Mol. Med. Rep. 2012, 5, 895–900. [Google Scholar] [CrossRef]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xu, L.; Rodriguez-Agudo, D.; Li, X.; Heuman, D.M.; Hylemon, P.B.; Pandak, W.M.; Ren, S. 25-Hydroxycholesterol-3-sulfate regulates macrophage lipid metabolism via the LXR/SREBP-1 signaling pathway. Am. J. Physiol. Metab. 2008, 295, E1369–E1379. [Google Scholar] [CrossRef]
- Wang, F.; Ma, Y.; Barrett, J.W.; Gao, X.; Loh, J.; Barton, E.; Virgin, H.W.; McFadden, G. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat. Immunol. 2004, 5, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e1011. [Google Scholar] [CrossRef] [PubMed]
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Doms, A.; Sanabria, T.; Hansen, J.N.; Altan-Bonnet, N.; Holm, G.H. 25-Hydroxycholesterol Production by the Cholesterol-25-Hydroxylase Interferon-Stimulated Gene Restricts Mammalian Reovirus Infection. J. Virol. 2018, 92, e01047-18. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef]
- Li, C.; Deng, Y.Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.Y.; Zhang, N.N.; Watanabe, M.; Dong, H.L.; Liu, P.; et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef]
- Ke, W.; Fang, L.; Jing, H.; Tao, R.; Wang, T.; Li, Y.; Long, S.; Wang, D.; Xiao, S. Cholesterol 25-Hydroxylase Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication through Enzyme Activity-Dependent and -Independent Mechanisms. J. Virol. 2017, 91, e00827-17. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, L.; Zhang, L.; Guo, Z.Z.; Lu, S.F.; Ming, S.L.; Li, G.L.; Wan, B.; Tian, K.G.; Yang, G.Y.; et al. Cholesterol 25-hydroxylase acts as a host restriction factor on pseudorabies virus replication. J. Gen. Virol. 2017, 98, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. S3-3—Oxysterols in the orchestra of liver cell metabolism. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, L.; Pandak, W.M.; Heuman, D.; Hylemon, P.B.; Ren, S. High Glucose Induces Lipid Accumulation via 25-Hydroxycholesterol DNA-CpG Methylation. iScience 2020, 23, 101102. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, K.; Kumar, S.M.; Clemens, D.L.; Dey, A. Inhibition of CYP2E1 leads to decreased advanced glycated end product formation in high glucose treated ADH and CYP2E1 over-expressing VL-17A cells. Biochim. Biophys. Acta 2013, 1830, 4407–4416. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Regulation of bile acid synthesis. Front. Biosci. 1998, 3, d176–d193. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Stravitz, R.T.; Vlahcevic, Z.R. Molecular genetics and regulation of bile acid biosynthesis. Prog. Liver Dis. 1994, 12, 99–120. [Google Scholar] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Gurley, E.C.; Stravitz, R.T.; Litz, J.S.; Pandak, W.M.; Chiang, J.Y.; Vlahcevic, Z.R. Hormonal regulation of cholesterol 7 alpha-hydroxylase mRNA levels and transcriptional activity in primary rat hepatocyte cultures. J. Biol. Chem. 1992, 267, 16866–16871. [Google Scholar]
- Pandak, W.M.; Schwarz, C.; Hylemon, P.B.; Mallonee, D.; Valerie, K.; Heuman, D.M.; Fisher, R.A.; Redford, K.; Vlahcevic, Z.R. Effects of CYP7A1 overexpression on cholesterol and bile acid homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G878–G889. [Google Scholar] [CrossRef]
- Stravitz, R.T.; Hylemon, P.B.; Heuman, D.M.; Hagey, L.R.; Schteingart, C.D.; Ton-Nu, H.T.; Hofmann, A.F.; Vlahcevic, Z.R. Transcriptional regulation of cholesterol 7 alpha-hydroxylase mRNA by conjugated bile acids in primary cultures of rat hepatocytes. J. Biol. Chem. 1993, 268, 13987–13993. [Google Scholar]
- Pandak, W.M.; Ren, S.; Marques, D.; Hall, E.; Redford, K.; Mallonee, D.; Bohdan, P.; Heuman, D.; Gil, G.; Hylemon, P. Transport of cholesterol into mitochondria is rate-limiting for bile acid synthesis via the alternative pathway in primary rat hepatocytes. J. Biol. Chem. 2002, 277, 48158–48164. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Reitz, J.; De Brabander, J.K.; Feramisco, J.D.; Li, L.; Brown, M.S.; Goldstein, J.L. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 2004, 279, 52772–52780. [Google Scholar] [CrossRef] [PubMed]
- Bellosta, S.; Corsini, A.; Bernini, F.; Granata, A.; Didoni, G.; Mazzotti, M.; Fumagalli, R. 27-Hydroxycholesterol modulation of low density lipoprotein metabolism in cultured human hepatic and extrahepatic cells. Feb. Lett. 1993, 332, 115–118. [Google Scholar] [CrossRef]
- Corsini, A.; Verri, D.; Raiteri, M.; Quarato, P.; Paoletti, R.; Fumagalli, R. Effects of 26-aminocholesterol, 27-hydroxycholesterol, and 25-hydroxycholesterol on proliferation and cholesterol homeostasis in arterial myocytes. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Diczfalusy, U. On the formation and possible biological role of 25-hydroxycholesterol. Biochimie 2013, 95, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Hylemon, P.B.; Marques, D.; Gurley, E.; Bodhan, P.; Hall, E.; Redford, K.; Gil, G.; Pandak, W.M. Overexpression of cholesterol transporter StAR increases in vivo rates of bile acid synthesis in the rat and mouse. Hepatology 2004, 40, 910–917. [Google Scholar] [CrossRef]
- Ren, S.; Li, X.; Rodriguez-Agudo, D.; Gil, G.; Hylemon, P.; Pandak, W.M. Sulfated oxysterol, 25HC3S, is a potent regulator of lipid metabolism in human hepatocytes. Lipids 2007, 360, 802–808. [Google Scholar] [CrossRef]
- Sánchez-Guijo, A.; Oji, V.; Hartmann, M.F.; Schuppe, H.C.; Traupe, H.; Wudy, S.A. High levels of oxysterol sulfates in serum of patients with steroid sulfatase deficiency. J. Lipid Res. 2015, 56, 403–412. [Google Scholar] [CrossRef]
- Töröcsik, D.; Szanto, A.; Nagy, L. Oxysterol signaling links cholesterol metabolism and inflammation via the liver X receptor in macrophages. Mol. Asp. Med. 2009, 30, 134–152. [Google Scholar] [CrossRef]
- Xu, L.; Kim, J.K.; Bai, Q.; Zhang, X.; Kakiyama, G.; Min, H.K.; Sanyal, A.J.; Pandak, W.M.; Ren, S. 5-cholesten-3beta,25-diol 3-sulfate decreases lipid accumulation in diet-induced nonalcoholic fatty liver disease mouse model. Mol. Pharmacol. 2013, 83, 648–658. [Google Scholar] [CrossRef]
- Xu, L.; Bai, Q.; Rodriguez-Agudo, D.; Hylemon, P.B.; Heuman, D.M.; Pandak, W.M.; Ren, S. Regulation of hepatocyte lipid metabolism and inflammatory response by 25-hydroxycholesterol and 25-hydroxycholesterol-3-sulfate. Lipids 2010, 45, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Xu, L.; Kakiyama, G.; Runge-Morris, M.A.; Hylemon, P.B.; Yin, L.; Pandak, W.M.; Ren, S. Sulfation of 25-hydroxycholesterol by SULT2B1b decreases cellular lipids via the LXR/SREBP-1c signaling pathway in human aortic endothelial cells. Atherosclerosis 2011, 214, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Ning, Y. Sulfation of 25-hydroxycholesterol regulates lipid metabolism, inflammatory responses, and cell proliferation. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E123–E130. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shen, S.; Ma, Y.; Kim, J.K.; Rodriguez-Agudo, D.; Heuman, D.M.; Hylemon, P.B.; Pandak, W.M.; Ren, S. 25-Hydroxycholesterol-3-sulfate attenuates inflammatory response via PPARgamma signaling in human THP-1 macrophages. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E788–E799. [Google Scholar] [CrossRef]
- Vanden Berghe, W.; Vermeulen, L.; Delerive, P.; De Bosscher, K.; Staels, B.; Haegeman, G. A paradigm for gene regulation: Inflammation, NF-kappaB and PPAR. Adv. Exp. Med. Biol. 2003, 544, 181–196. [Google Scholar]
- Ning, Y.; Kim, J.K.; Min, H.K.; Ren, S. Cholesterol metabolites alleviate injured liver function and decrease mortality in an LPS-induced mouse model. Metab. Clin. Exp. 2017, 71, 83–93. [Google Scholar] [CrossRef]
- Yu, W.; McIntosh, C.; Lister, R.; Zhu, I.; Han, Y.; Ren, J.; Landsman, D.; Lee, E.; Briones, V.; Terashima, M.; et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014, 24, 1613–1623. [Google Scholar] [CrossRef]
- Castellano-Castillo, D.; Moreno-Indias, I.; Sanchez-Alcoholado, L.; Ramos-Molina, B.; Alcaide-Torres, J.; Morcillo, S.; Ocaña-Wilhelmi, L.; Tinahones, F.; Queipo-Ortuño, M.I.; Cardona, F. Altered Adipose Tissue DNA Methylation Status in Metabolic Syndrome: Relationships Between Global DNA Methylation and Specific Methylation at Adipogenic, Lipid Metabolism and Inflammatory Candidate Genes and Metabolic Variables. J. Clin. Med. 2019, 8, 87. [Google Scholar] [CrossRef]
- Gowher, H.; Jeltsch, A. Mammalian DNA methyltransferases: New discoveries and open questions. Biochem. Soc. Trans. 2018, 46, 1191–1202. [Google Scholar] [CrossRef]
- Ren, W.; Gao, L.; Song, J. Structural Basis of DNMT1 and DNMT3A-Mediated DNA Methylation. Genes 2018, 9, 620. [Google Scholar] [CrossRef]
- Canbay, A.; Bechmann, L.; Gerken, G. Lipid metabolism in the liver. Z. Fur. Gastroenterol. 2007, 45, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr. Diabetes Rep. 2006, 6, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Stojsavljević, S.; Gomerčić Palčić, M.; Virović Jukić, L.; Smirčić Duvnjak, L.; Duvnjak, M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 18070–18091. [Google Scholar] [CrossRef]
- du Plessis, J.; Korf, H.; van Pelt, J.; Windmolders, P.; Vander Elst, I.; Verrijken, A.; Hubens, G.; Van Gaal, L.; Cassiman, D.; Nevens, F.; et al. Pro-Inflammatory Cytokines but Not Endotoxin-Related Parameters Associate with Disease Severity in Patients with NAFLD. PLoS ONE 2016, 11, e0166048. [Google Scholar] [CrossRef]
- Kumar, R.; Prakash, S.; Chhabra, S.; Singla, V.; Madan, K.; Gupta, S.D.; Panda, S.K.; Khanal, S.; Acharya, S.K. Association of pro-inflammatory cytokines, adipokines & oxidative stress with insulin resistance & non-alcoholic fatty liver disease. Indian J. Med. Res. 2012, 136, 229–236. [Google Scholar]
- Kemp, W.S.J.; Kim, M.J.; Ellis, D.; Brown, J.; Theeuwes, F.; Lin, W.Q. Safety and pharmacokinetics of DUR-928 in patients with non-alcoholic steatohepatitis—A Phase 1b study. In Proceedings of the Poster Session Presented at The International Liver Congress, Amsterdam, The Netherlands, 19–23 April 2017. [Google Scholar]
- Shi, X.; Cheng, Q.; Xu, L.; Yan, J.; Jiang, M.; He, J.; Xu, M.; Stefanovic-Racic, M.; Sipula, I.; O’Doherty, R.M.; et al. Cholesterol sulfate and cholesterol sulfotransferase inhibit gluconeogenesis by targeting hepatocyte nuclear factor 4alpha. Mol. Cell. Biol. 2014, 34, 485–497. [Google Scholar] [CrossRef]
- Bi, Y.; Shi, X.; Zhu, J.; Guan, X.; Garbacz, W.G.; Huang, Y.; Gao, L.; Yan, J.; Xu, M.; Ren, S.; et al. Regulation of Cholesterol Sulfotransferase SULT2B1b by HNF4alpha Constitutes a Negative Feedback Control of Hepatic Gluconeogenesis. Mol. Cell. Biol. 2018, 38, e00654-17. [Google Scholar] [CrossRef]
- Forbes, S.J.; Newsome, P.N. Liver regeneration—Mechanisms and models to clinical application. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bai, Q.; Kakiyama, G.; Xu, L.; Kim, J.K.; Pandak, W.M., Jr.; Ren, S. Cholesterol metabolite, 5-cholesten-3beta-25-diol-3-sulfate, promotes hepatic proliferation in mice. J. Steroid Biochem. Mol. Biol. 2012, 132, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bai, Q.; Xu, L.; Kakiyama, G.; Pandak, W.M., Jr.; Zhang, Z.; Ren, S. Cytosolic sulfotransferase 2B1b promotes hepatocyte proliferation gene expression in vivo and in vitro. Am. J. Physiol. Endocrinol. Metab. 2012, 303, G344–G355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- DePass, L.R.S.J.; Miksztal, A.; Lin, W. In vivo tissue distribution and elimination of DUR-928, a first in class therapeutic for treatment of hepatic and renal disease. In Proceedings of the Poster Session Presented at the Society of Toxicology Annual Meeting, San Antonio, TX, USA, 11–15 March 2018. [Google Scholar]
- Shah, J.E.D.; Miksztal, A.; Lin, W.Q. Safety and single ascending dose pharmacokinetic study of DUR-928 in patients with chronic kidney disease versus matched control subjects. In Proceedings of the Poster Session Presented at the American Society of Nephrology annual Kidney Week Meeting, San Diego, CA, USA, 23–28 October 2018. [Google Scholar]
- Hassanein, T. Safety and efficacy of DUR-928: A potential new therapy for acute alcoholic hepatitis. In Proceedings of the AASLD the Liver Meeting®, Boston, MA, USA, 12 November 2019. [Google Scholar]
- Packard, C.J. LDL cholesterol: How low to go? Trends Cardiovasc. Med. 2018, 28, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Parhofer, K.G. New approaches to address dyslipidemia. Curr. Opin. Lipidol. 2017, 28, 452–457. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, X.; Ren, S. Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage. Metabolites 2021, 11, 9. https://doi.org/10.3390/metabo11010009
Wang Y, Li X, Ren S. Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage. Metabolites. 2021; 11(1):9. https://doi.org/10.3390/metabo11010009
Chicago/Turabian StyleWang, Yaping, Xiaobo Li, and Shunlin Ren. 2021. "Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage" Metabolites 11, no. 1: 9. https://doi.org/10.3390/metabo11010009
APA StyleWang, Y., Li, X., & Ren, S. (2021). Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage. Metabolites, 11(1), 9. https://doi.org/10.3390/metabo11010009