Lipid Players of Cellular Senescence


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
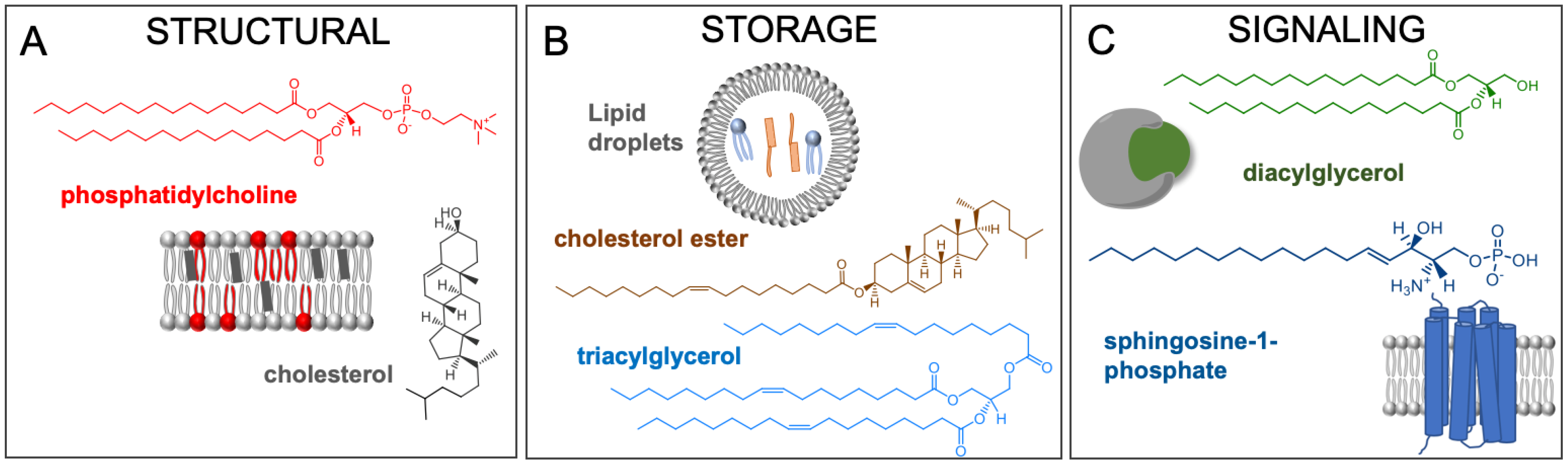
Lipid Diversity and Function
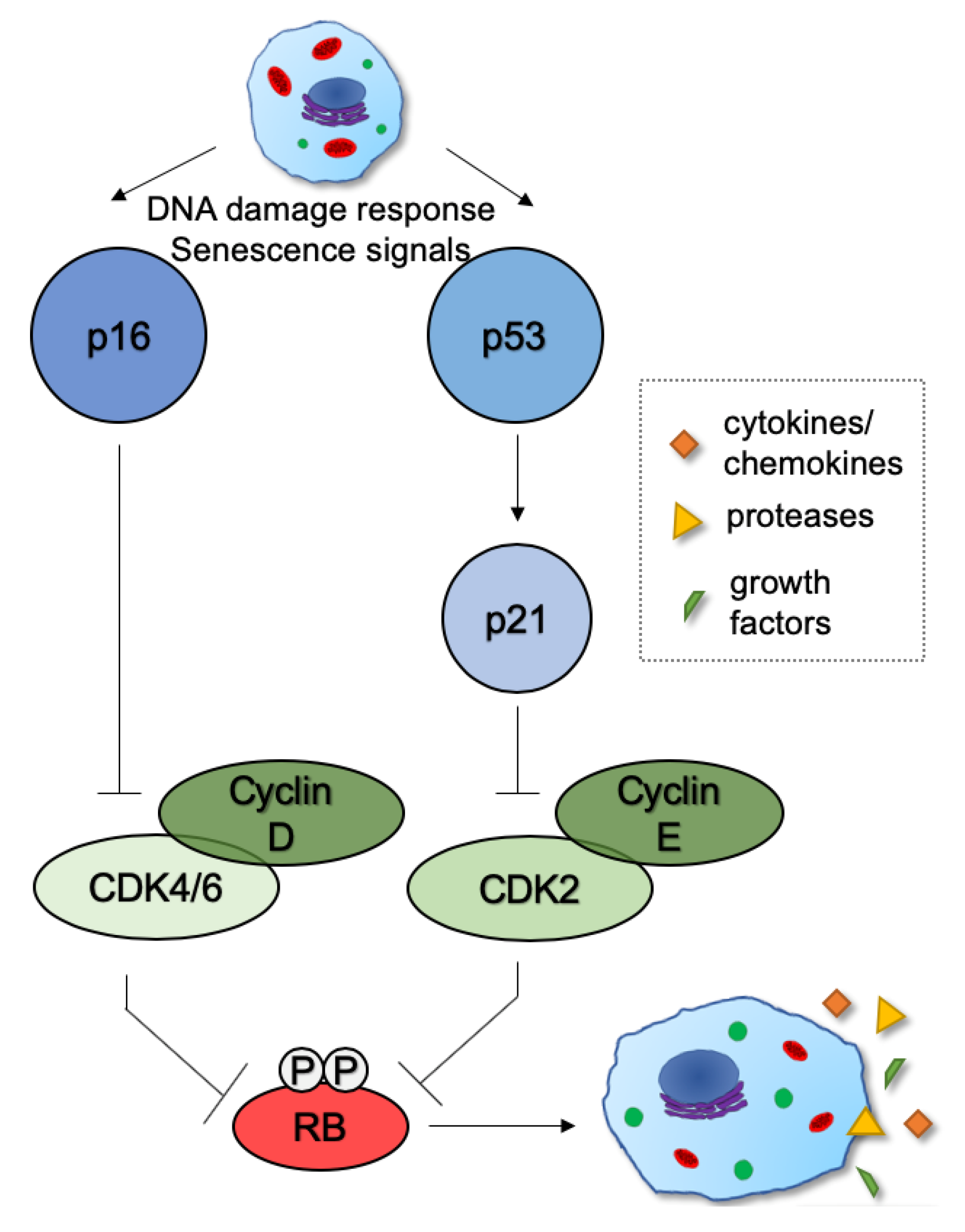
2. Mechanisms and Functions of Cellular Senescence
3. Various Lipid Classes Have Been Linked to Senescence
3.1. Fatty Acids
3.2. CD36
3.3. Glycerolipids
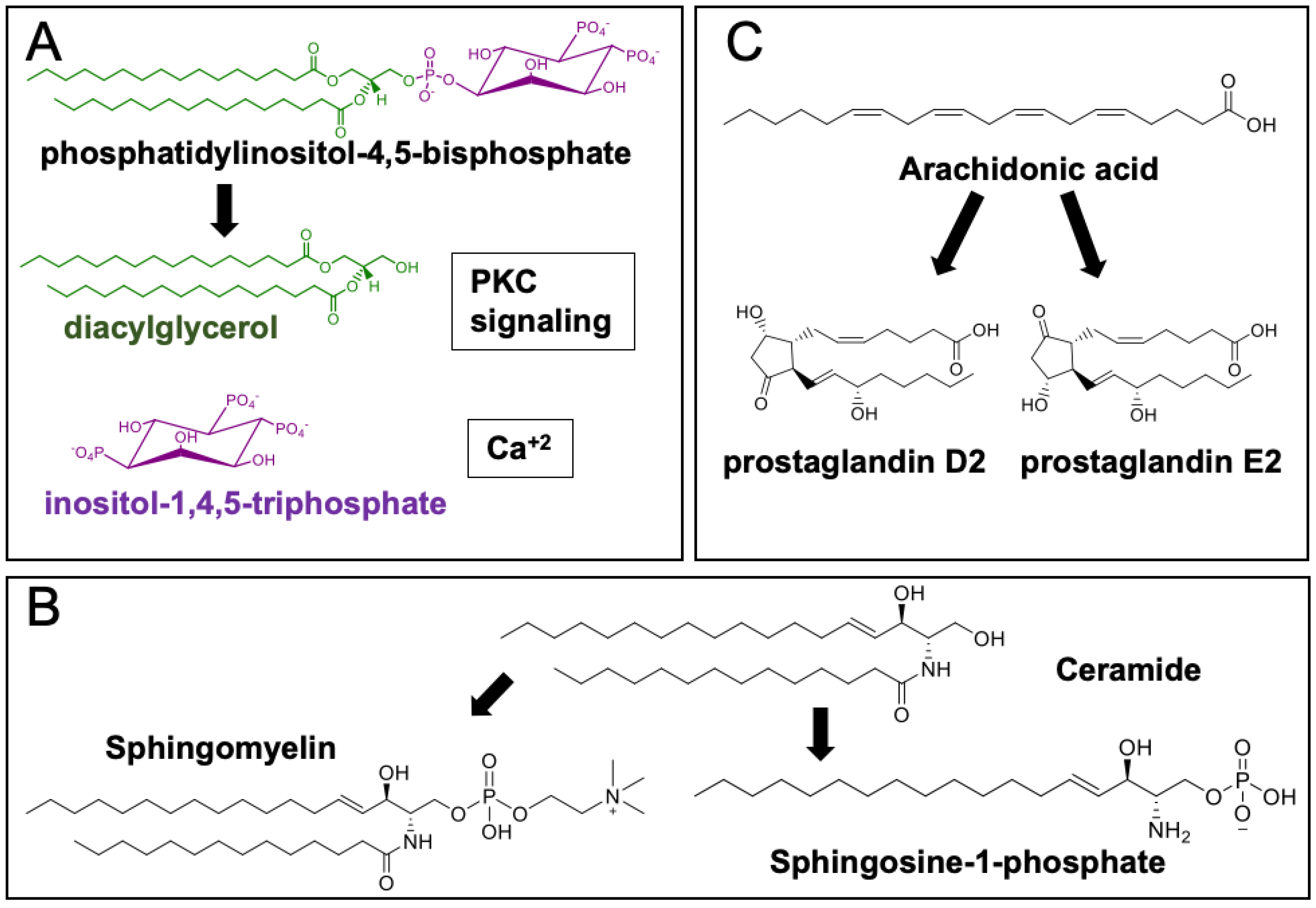
3.4. Phospholipids
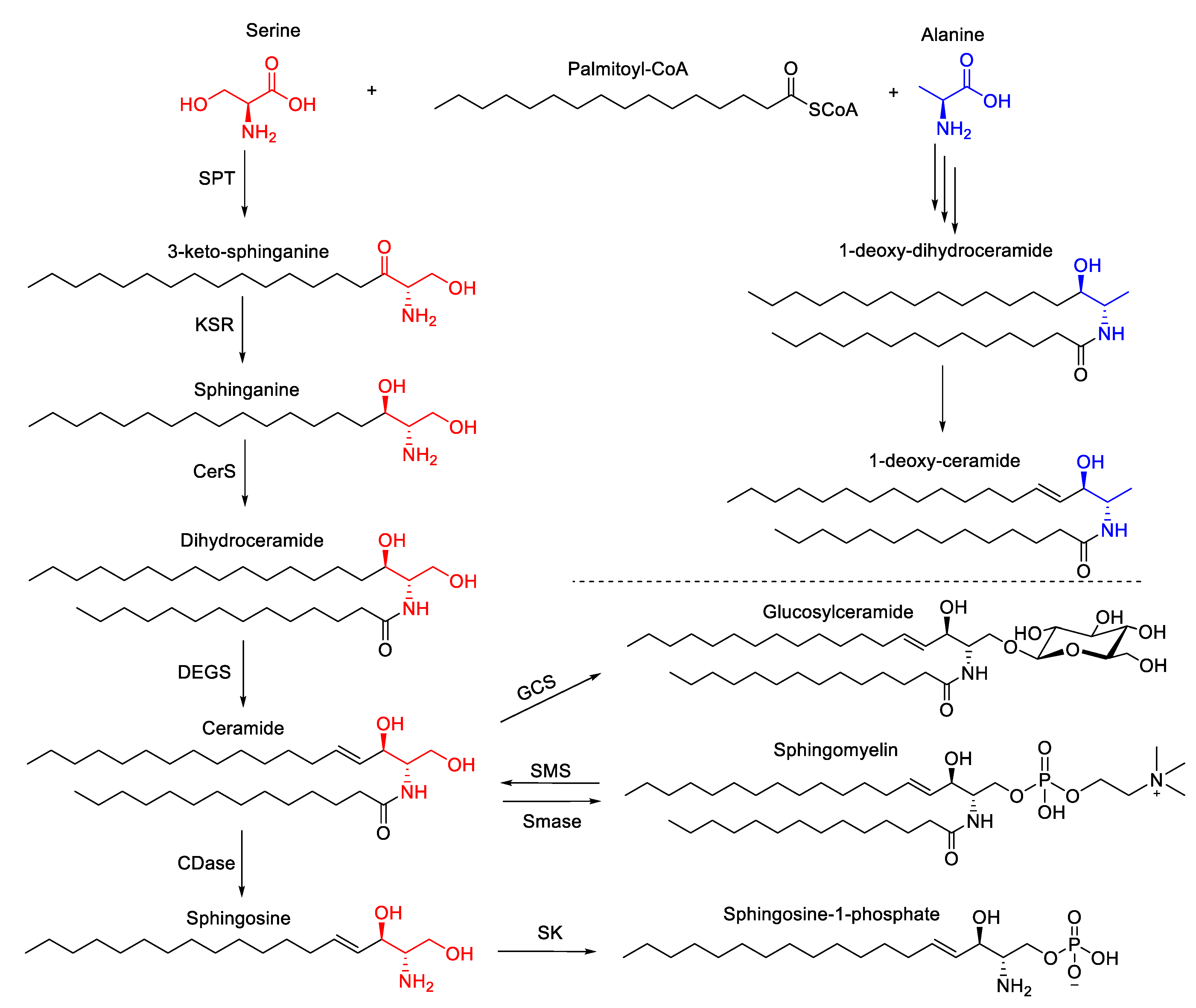
3.5. Sphingolipids
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Fahy, E.; Cotter, D.; Sud, M.; Subramaniam, S. Lipid classification, structures and tools. Biochim. Biophys. Acta 2011, 1811, 637–647. [Google Scholar] [CrossRef]
- Parker, B.L.; Calkin, A.C.; Seldin, M.M.; Keating, M.F.; Tarling, E.J.; Yang, P.; Moody, S.C.; Liu, Y.; Zerenturk, E.J.; Needham, E.J.; et al. An integrative systems genetic analysis of mammalian lipid metabolism. Nature 2019, 567, 187–193. [Google Scholar]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Yoshida, K.; Nagatoishi, S.; Kuroda, D.; Suzuki, N.; Murata, T.; Tsumoto, K. Phospholipid Membrane Fluidity Alters Ligand Binding Activity of a G Protein-Coupled Receptor by Shifting the Conformational Equilibrium. Biochemistry 2019, 58, 504–508. [Google Scholar] [CrossRef]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 2004, 1666, 62–87. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2018, 20, 137–155. [Google Scholar] [CrossRef]
- Newton, A.C. Lipid activation of protein kinases. J. Lipid Res. 2009, 50, S266–S271. [Google Scholar] [CrossRef]
- Spitaler, M.; Cantrell, D.A. Protein kinase C and beyond. Nat. Immunol. 2004, 5, 785–790. [Google Scholar] [CrossRef]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef]
- Gallegos, L.L.; Kunkel, M.T.; Newton, A.C. Targeting Protein Kinase C Activity Reporter to Discrete Intracellular Regions Reveals Spatiotemporal Differences in Agonist-dependent Signaling. J. Biol. Chem. 2006, 281, 30947–30956. [Google Scholar] [CrossRef]
- Medkova, M.; Cho, W. Differential membrane-binding and activation mechanisms of protein kinase C-alpha and -epsilon. Biochemistry 1998, 37, 4892–4900. [Google Scholar]
- Igumenova, T.I. Dynamics and Membrane Interactions of Protein Kinase C. Biochemistry 2015, 54, 4953–4968. [Google Scholar] [CrossRef]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Palaniyandi, S.S.; Sun, L.; Ferreira, J.C.; Mochly-Rosen, D. Protein kinase C in heart failure: A therapeutic target? Cardiovasc. Res. 2009, 82, 229–239. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Author Correction: Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 673. [Google Scholar] [CrossRef]
- Taha, T.A.; Mullen, T.D.; Obeid, L.M. A house divided: Ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta 2006, 1758, 2027–2036. [Google Scholar] [CrossRef]
- Calise, S.; Blescia, S.; Cencetti, F.; Bernacchioni, C.; Donati, C.; Bruni, P. Sphingosine 1-phosphate stimulates proliferation and migration of satellite cells. Biochim. Biophys. Acta 2012, 1823, 439–450. [Google Scholar] [CrossRef]
- Kimura, T.; Watanabe, T.; Sato, K.; Kon, J.; Tomura, H.; Tamama, K.; Kuwabara, A.; Kanda, T.; Kobayashi, I.; Ohta, H.; et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem. J. 2000, 348 Pt 1, 71–76. [Google Scholar]
- Hla, T.; Dannenberg, A.J. Sphingolipid Signaling in Metabolic Disorders. Cell Metab. 2012, 16, 420–434. [Google Scholar] [CrossRef]
- Pereira, P.A.T.; Assis, P.A.; Prado, M.K.B.; Ramos, S.G.; Aronoff, D.M.; Silva, F.W.G.P.; Sorgi, C.A.; Faccioli, L.H. Prostaglandins D2 and E2 have opposite effects on alveolar macrophages infected with Histoplasma capsulatum. J. Lipid Res. 2017, 59, 195–206. [Google Scholar] [CrossRef]
- Storck, E.M.; Özbalci, C.; Eggert, U.S. Lipid Cell Biology: A Focus on Lipids in Cell Division. Annu. Rev. Biochem. 2018, 87, 839–869. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef]
- Agmon, E.; Stockwell, B.R. Lipid homeostasis and regulated cell death. Curr. Opin. Chem. Biol. 2017, 39, 83–89. [Google Scholar] [CrossRef]
- Shaw, J.; Costa-Pinheiro, P.; Patterson, L.; Drews, K.; Spiegel, S.; Kester, M. Novel Sphingolipid-Based Cancer Therapeutics in the Personalized Medicine Era. Adv. Cancer Res. 2018, 140, 327–366. [Google Scholar] [CrossRef]
- Johnson, A.A.; Stolzing, A. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell 2019, 18, e13048. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; DeMaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Saitou, M.; Lizardo, D.Y.; Taskent, R.O.; Millner, A.; Gokcumen, O.; Atilla-Gokcumen, G.E.; Lizardo, D. An evolutionary transcriptomics approach links CD36 to membrane remodeling in replicative senescence. Mol. Omics 2018, 14, 237–246. [Google Scholar] [CrossRef]
- Millner, A.; Lizardo, D.Y.; Atilla-Gokcumen, G.E. Untargeted Lipidomics Highlight the Depletion of Deoxyceramides during Therapy-Induced Senescence. Proteomics 2020, 20, e2000013. [Google Scholar] [CrossRef]
- Lizardo, D.Y.; Lin, Y.-L.; Gokcumen, O.; Atilla-Gokcumen, G.E. Regulation of lipids is central to replicative senescence. Mol. BioSyst. 2017, 13, 498–509. [Google Scholar] [CrossRef]
- Chong, M.; Yin, T.; Chen, R.; Xiang, H.; Yuan, L.; Ding, Y.; Pan, C.C.; Tang, Z.; Alexander, P.B.; Li, Q.; et al. CD 36 initiates the secretory phenotype during the establishment of cellular senescence. EMBO Rep. 2018, 19, e45274. [Google Scholar] [CrossRef]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A Senescence Program Controlled by p53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Toussaint, O.; Royer, V.; Salmon, M.; Remacle, J. Stress-induced premature senescence and tissue ageing. Biochem. Pharmacol. 2002, 64, 1007–1009. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar]
- Druelle, C.; Drullion, C.; Deslé, J.; Martin, N.; Saas, L.; Cormenier, J.; Malaquin, N.; Huot, L.; Slomianny, C.; Bouali, F.; et al. ATF6α regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 2016, 7, 67699–67715. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Maciel-Barón, L.A.; Morales-Rosales, S.L.; Aquino-Cruz, A.A.; Triana-Martinez, F.; Galván-Arzate, S.; Luna-López, A.; González-Puertos, V.Y.; López-Díazguerrero, N.E.; Torres, C.; Königsberg, M. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. AGE 2016, 38, 26. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; Van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- DeMaria, M.; Ohtani, N.; Youssef, S.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.-M.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef]
- Monnerie, S.; Comte, B.; Ziegler, D.; Morais, J.A.; Pujos-Guillot, E.; Gaudreau, P. Metabolomic and Lipidomic Signatures of Metabolic Syndrome and its Physiological Components in Adults: A Systematic Review. Sci. Rep. 2020, 10, 669. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Single Mol. Single Cell Seq. 2017, 1043, 227–256. [Google Scholar] [CrossRef]
- Parrinello, S.; Coppé, J.-P.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118 Pt 3, 485–496. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Awad, P.; Campisi, J.; Desprez, P.-Y. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2012, 5, 39–44. [Google Scholar] [CrossRef]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Zdanov, S.; Bernard, D.; Debacqchainiaux, F.; Martien, S.; Gosselin, K.; Vercamer, C.; Chelli, F.; Toussaint, O.; Abbadie, C. Normal or stress-induced fibroblast senescence involves COX-2 activity. Exp. Cell Res. 2007, 313, 3046–3056. [Google Scholar] [CrossRef]
- Chang, B.-D.; Swift, M.E.; Shen, M.; Fang, J.; Broude, E.V.; Roninson, I.B. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc. Natl. Acad. Sci. USA 2001, 99, 389–394. [Google Scholar] [CrossRef]
- Huang, N.N.; Wang, D.J.; Heppel, L.A. Stimulation of aged human lung fibroblasts by extracellular ATP via suppression of arachidonate metabolism. J. Biol. Chem. 1993, 268, 10789–10795. [Google Scholar]
- Chou, J.P.; Ramirez, C.M.; Ryba, D.M.; Koduri, M.P.; Effros, R.B. Prostaglandin E2 Promotes Features of Replicative Senescence in Chronically Activated Human CD8+ T Cells. PLoS ONE 2014, 9, e99432. [Google Scholar] [CrossRef]
- Schroeder, E.A.; Brunet, A. Lipid Profiles and Signals for Long Life. Trends Endocrinol. Metab. 2015, 26, 589–592. [Google Scholar] [CrossRef]
- Pararasa, C.; Ikwuobe, J.; Shigdar, S.; Boukouvalas, A.; Nabney, I.T.; Brown, J.E.; Devitt, A.; Bailey, C.J.; Bennett, S.J.; Griffiths, H.R. Age-associated changes in long-chain fatty acid profile during healthy aging promote pro-inflammatory monocyte polarization via PPARgamma. Aging Cell 2016, 15, 128–139. [Google Scholar]
- Labora, J.A.F.; Carpintero-Fernández, P.; Jordan, S.J.D.; Shikh-Bahaei, T.; Abdullah, S.M.; Mahenthiran, M.; Rodríguez-Navarro, J.A.; Niklison-Chirou, M.V.; O’Loghlen, A. FASN activity is important for the initial stages of the induction of senescence. Cell Death Dis. 2019, 10, 318. [Google Scholar] [CrossRef]
- Marmisolle, I.; Martínez, J.; Liu, J.; Mastrogiovanni, M.; Fergusson, M.M.; Rovira, I.I.; Castro, L.; Trostchansky, A.; Moreno, M.; Cao, L.; et al. Reciprocal regulation of acetyl-CoA carboxylase 1 and senescence in human fibroblasts involves oxidant mediated p38 MAPK activation. Arch. Biochem. Biophys. 2017, 613, 12–22. [Google Scholar] [CrossRef]
- Fritz, I.B.; Yue, K.T. Long-chain carnitine acyltransferase and the role of acylcarnitine derivatives in the catalytic increase of fatty acid oxidation induced by carnitine. J. Lipid Res. 1963, 4, 279–288. [Google Scholar]
- Stephens, F.B.; Constantin-Teodosiu, D.; Greenhaff, P. New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. J. Physiol. 2007, 581 Pt 2, 431–444. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Huang, Y.; Zeng, H.; Hu, B.; Guan, L.; Zhang, H.; Yu, A.M.; Johnson, C.H.; Gonzalez, F.J.; et al. PPARalpha regulates tumor cell proliferation and senescence via a novel target gene carnitine palmitoyltransferase 1C. Carcinogenesis 2017, 38, 474–483. [Google Scholar]
- Wakil, S.J.; Abu-Elheiga, L. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2008, 50, S138–S143. [Google Scholar] [CrossRef]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar]
- Seok, J.; Jung, H.S.; Park, S.; Lee, J.O.; Kim, C.J.; Kim, G.J. Alteration of fatty acid oxidation by increased CPT1A on replicative senescence of placenta-derived mesenchymal stem cells. Stem Cell Res. Ther. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-Function of CD36 and Importance of Fatty Acid Signal Transduction in Fat Metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef]
- Smith, J.; Su, X.; El-Maghrabi, R.; Stahl, P.D.; Abumrad, N.A. Opposite Regulation of CD36 Ubiquitination by Fatty Acids and Insulin: Effects on fatty acid uptake. J. Biol. Chem. 2008, 283, 13578–13585. [Google Scholar] [CrossRef]
- Hoosdally, S.J.; Andress, E.J.; Wooding, C.; Martin, C.A.; Linton, K.J. The Human Scavenger Receptor CD36: Glycosylation status and its role in trafficking and function. J. Biol. Chem. 2009, 284, 16277–16288. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef]
- Luiken, J.J.F.P.; Chanda, D.; Nabben, M.; Neumann, D.; Glatz, J.F.C. Post-translational modifications of CD36 (SR-B2): Implications for regulation of myocellular fatty acid uptake. Biochim. Biophys. Acta 2016, 1862, 2253–2258. [Google Scholar] [CrossRef]
- Coburn, C.T.; Knapp, F.F., Jr.; Febbraio, M.; Beets, A.L.; Silverstein, R.L.; Abumrad, N.A. Defective Uptake and Utilization of Long Chain Fatty Acids in Muscle and Adipose Tissues of CD36 Knockout Mice. J. Biol. Chem. 2000, 275, 32523–32529. [Google Scholar] [CrossRef]
- Nozaki, S.; Tanaka, T.; Yamashita, S.; Sohmiya, K.; Yoshizumi, T.; Okamoto, F.; Kitaura, Y.; Kotake, C.; Nishida, H.; Nakata, A.; et al. CD36 mediates long-chain fatty acid transport in human myocardium: Complete myocardial accumulation defect of radiolabeled long-chain fatty acid analog in subjects with CD36 deficiency. Mol. Cell. Biochem. 1999, 192, 129–135. [Google Scholar]
- Son, N.-H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.-Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a Scavenger Receptor Involved in Immunity, Metabolism, Angiogenesis, and Behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Gugiu, B.; Fox, P.L.; et al. Identification of a Novel Family of Oxidized Phospholipids That Serve as Ligands for the Macrophage Scavenger Receptor CD36. J. Biol. Chem. 2002, 277, 38503–38516. [Google Scholar] [CrossRef]
- Jimenez-Dalmaroni, M.J.; Xiao, N.; Corper, A.L.; Verdino, P.; Ainge, G.D.; Larsen, D.S.; Painter, G.F.; Rudd, P.M.; Dwek, R.A.; Hoebe, K.; et al. Soluble CD36 Ectodomain Binds Negatively Charged Diacylglycerol Ligands and Acts as a Co-Receptor for TLR2. PLoS ONE 2009, 4, e7411. [Google Scholar] [CrossRef]
- Calvo, D.; Gómez-Coronado, D.; Suárez, Y.; Lasunción, M.A.; Vega, M.A. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J. Lipid Res. 1998, 39, 777–788. [Google Scholar]
- Dawson, D.W.; Pearce, S.F.A.; Zhong, R.; Silverstein, R.L.; Frazier, W.A.; Bouck, N. CD36 Mediates the In Vitro Inhibitory Effects of Thrombospondin-1 on Endothelial Cells. J. Cell Biol. 1997, 138, 707–717. [Google Scholar] [CrossRef]
- Pennathur, S.; Pasichnyk, K.; Bahrami, N.M.; Zeng, L.; Febbraio, M.; Yamaguchi, I.; Okamura, D.M. The macrophage phagocytic receptor CD36 promotes fibrogenic pathways on removal of apoptotic cells during chronic kidney injury. Am. J. Pathol. 2015, 185, 2232–2245. [Google Scholar] [CrossRef]
- Tandon, N.N.; Kralisz, U.; Jamieson, G.A. Identification of glycoprotein IV (CD36) as a primary receptor for platelet-collagen adhesion. J. Biol. Chem. 1989, 264, 7576–7583. [Google Scholar]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El Khoury, J. CD36, a Class B Scavenger Receptor, Is Expressed on Microglia in Alzheimer’s Disease Brains and Can Mediate Production of Reactive Oxygen Species in Response to β-Amyloid Fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Kuchibhotla, S.; Westfall, K.M.; Silverstein, R.L.; Morton, R.E.; Febbraio, M. A CD36-dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc. Res. 2011, 89, 604–613. [Google Scholar] [CrossRef]
- Coleman, R.A.; Mashek, D.G. Mammalian Triacylglycerol Metabolism: Synthesis, Lipolysis, and Signaling. Chem. Rev. 2011, 111, 6359–6386. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef]
- Boren, J.; Brindle, K.M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef]
- Li, N.; Lizardo, D.Y.; Atilla-Gokcumen, G.E. Specific Triacylglycerols Accumulate via Increased Lipogenesis During 5-FU-Induced Apoptosis. ACS Chem. Biol. 2016, 11, 2583–2587. [Google Scholar] [CrossRef]
- Sakuragi, T.; Kosako, H.; Nagata, S. Phosphorylation-mediated activation of mouse Xkr8 scramblase for phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA 2019, 116, 2907–2912. [Google Scholar] [CrossRef]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016, 23, 952–961. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef]
- Rivera, R.; Chun, J. Biological effects of lysophospholipids. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 25–46. [Google Scholar] [CrossRef]
- Konno, T.; Kotani, T.; Setiawan, J.; Nishigaito, Y.; Sawada, N.; Imada, S.; Saito, Y.; Murata, Y.; Matozaki, T. Role of lysophosphatidic acid in proliferation and differentiation of intestinal epithelial cells. PLoS ONE 2019, 14, e0215255. [Google Scholar] [CrossRef]
- Weiner, J.A.; Chun, J. Schwann cell survival mediated by the signaling phospholipid lysophosphatidic acid. Proc. Natl. Acad. Sci. USA 1999, 96, 5233–5238. [Google Scholar] [CrossRef]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef]
- Kostić, I.; Carvalho, I.F.; Aday, S.; Vazão, H.; Carvalheiro, T.; Grãos, M.; Duarte, A.; Cardoso, C.M.; Gonçalves, L.; Carvalho, L.; et al. Lysophosphatidic acid enhances survival of human CD34+ cells in ischemic conditions. Sci. Rep. 2015, 5, 16406. [Google Scholar] [CrossRef]
- Kano, K.; Matsumoto, H.; Inoue, A.; Yukiura, H.; Kanai, M.; Chun, J.; Ishii, S.; Shimizu, T.; Aoki, J. Molecular mechanism of lysophosphatidic acid-induced hypertensive response. Sci. Rep. 2019, 9, 2662. [Google Scholar] [CrossRef]
- Colles, S.M.; Chisolm, G.M. Lysophosphatidylcholine-induced cellular injury in cultured fibroblasts involves oxidative events. J. Lipid Res. 2000, 41, 1188–1198. [Google Scholar]
- Bansal, P.; Gaur, S.N.; Arora, N. Lysophosphatidylcholine plays critical role in allergic airway disease manifestation. Sci. Rep. 2016, 6, 27430. [Google Scholar] [CrossRef]
- Kakisaka, K.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Gores, G.J. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am. J. Physiol. Gastrointerest. Liver Physiol. 2012, 302, G77–G84. [Google Scholar] [CrossRef]
- Flor, A.C.; Doshi, A.P.; Kron, S.J. Modulation of therapy-induced senescence by reactive lipid aldehydes. Cell Death Discov. 2016, 2, 16045. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef]
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta 2002, 1585, 153–162. [Google Scholar] [CrossRef]
- Zhang, H.; Desai, N.N.; Olivera, A.; Seki, T.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 1991, 114, 155–167. [Google Scholar] [CrossRef]
- Lange, Y.; Swaisgood, M.H.; Ramos, B.V.; Steck, T.L. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J. Biol. Chem. 1989, 264, 3786–3793. [Google Scholar]
- Klenk, H.-D.; Choppin, P.W. Glycosphingolipids of Plasma Membranes of Cultured Cells and an Enveloped Virus (SV5) Grown in These Cells. Proc. Natl. Acad. Sci. USA 1970, 66, 57–64. [Google Scholar] [CrossRef]
- Lönnfors, M.; Doux, J.P.; Killian, J.A.; Nyholm, T.K.; Slotte, J.P. Sterols Have Higher Affinity for Sphingomyelin than for Phosphatidylcholine Bilayers even at Equal Acyl-Chain Order. Biophys. J. 2011, 100, 2633–2641. [Google Scholar] [CrossRef]
- Gupta, A.K.; Rudney, H. Plasma membrane sphingomyelin and the regulation of HMG-CoA reductase activity and cholesterol biosynthesis in cell cultures. J. Lipid Res. 1991, 32, 125–136. [Google Scholar]
- Contreras, F.-X.; Ernst, A.M.; Haberkant, P.; Björkholm, P.; Lindahl, E.; Gönen, B.; Tischer, C.; Elofsson, A.; Von Heijne, G.; Thiele, C.; et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 2012, 481, 525–529. [Google Scholar] [CrossRef]
- Singh, P.; Chattopadhyay, A. Removal of sphingomyelin headgroup inhibits the ligand binding function of hippocampal serotonin1A receptors. Biochem. Biophys. Res. Commun. 2012, 419, 321–325. [Google Scholar] [CrossRef]
- Patterson, G.H.; Hirschberg, K.; Polishchuk, R.S.; Gerlich, D.; Phair, R.D.; Lippincott-Schwartz, J. Transport through the Golgi Apparatus by Rapid Partitioning within a Two-Phase Membrane System. Cell 2008, 133, 1055–1067. [Google Scholar] [CrossRef]
- Wojtal, K.A.; de Vries, E.; Hoekstra, D.; van Ijzendoorn, S.C. Efficient trafficking of MDR1/P-glycoprotein to apical canalicular plasma membranes in HepG2 cells requires PKA-RIIalpha anchoring and glucosylceramide. Mol. Biol. Cell. 2006, 17, 3638–3650. [Google Scholar]
- Iwabuchi, K.; Prinetti, A.; Sonnino, S.; Mauri, L.; Kobayashi, T.; Ishii, K.; Kaga, N.; Murayama, K.; Kurihara, H.; Nakayama, H.; et al. Involvement of very long fatty acid-containing lactosylceramide in lactosylceramide-mediated superoxide generation and migration in neutrophils. Glycoconj. J. 2008, 25, 355–356. [Google Scholar] [CrossRef][Green Version]
- Won, J.-S.; Singh, A.K.; Singh, I. Lactosylceramide: A lipid second messenger in neuroinflammatory disease. J. Neurochem. 2007, 103 (Suppl. 1), 180–191. [Google Scholar] [CrossRef]
- Venable, M.E.; Yin, X. Ceramide induces endothelial cell senescence. Cell Biochem. Funct. 2009, 27, 547–551. [Google Scholar] [CrossRef]
- Khayrullin, A.; Krishnan, P.; Martinez-Nater, L.; Mendhe, B.; Fulzele, S.; Liu, Y.; Mattison, J.A.; Hamrick, M.W. Very Long-Chain C24:1 Ceramide Is Increased in Serum Extracellular Vesicles with Aging and Can Induce Senescence in Bone-Derived Mesenchymal Stem Cells. Cells 2019, 8, 37. [Google Scholar] [CrossRef]
- Venable, M.E.; Lee, J.Y.; Smyth, M.J.; Bielawska, A.; Obeid, L.M. Role of Ceramide in Cellular Senescence. J. Biol. Chem. 1995, 270, 30701–30708. [Google Scholar] [CrossRef]
- Mouton, R.E.; Venable, M.E. Ceramide induces expression of the senescence histochemical marker, beta-galactosidase, in human fibroblasts. Mech. Ageing Dev. 2000, 113, 169–181. [Google Scholar]
- Kim, M.K.; Lee, W.; Yoon, G.-H.; Chang, E.-J.; Choi, S.-C.; Kim, S.W. Links between accelerated replicative cellular senescence and down-regulation of SPHK1 transcription. BMB Rep. 2019, 52, 220–225. [Google Scholar] [CrossRef]
- Heffernan-Stroud, L.A.; Helke, K.L.; Jenkins, R.W.; De Costa, A.-M.; Hannun, Y.A.; Obeid, L.M. Defining a role for sphingosine kinase 1 in p53-dependent tumors. Oncogene 2012, 31, 1166–1175. [Google Scholar] [CrossRef]
- Trayssac, M.; Hannun, Y.A.; Obeid, L.M. Role of sphingolipids in senescence: Implication in aging and age-related diseases. J. Clin. Investig. 2018, 128, 2702–2712. [Google Scholar] [CrossRef]
- Selvam, S.P.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58. [Google Scholar] [CrossRef]
- Adada, M.; Canals, D.; Hannun, Y.A.; Obeid, L.M. Sphingosine-1-phosphate receptor 2. FEBS J. 2013, 280, 6354–6366. [Google Scholar]
- Marchesini, N.; Osta, W.; Bielawski, J.; Luberto, C.; Obeid, L.M.; Hannun, Y.A. Role for Mammalian Neutral Sphingomyelinase 2 in Confluence-induced Growth Arrest of MCF7 Cells. J. Biol. Chem. 2004, 279, 25101–25111. [Google Scholar] [CrossRef]
- Jensen, J.-M.; Förl, M.; Winoto-Morbach, S.; Seite, S.; Schunck, M.; Proksch, E.; Schütze, S. Acid and neutral sphingomyelinase, ceramide synthase, and acid ceramidase activities in cutaneous aging. Exp. Dermatol. 2005, 14, 609–618. [Google Scholar] [CrossRef]
- Hitomi, K.; Okada, R.; Loo, T.M.; Miyata, K.; Nakamura, A.J.; Takahashi, A. DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses. Int. J. Mol. Sci. 2020, 21, 3720. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Single Mol. Single Cell Seq. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Jiménez-Rojo, N.; Sot, J.; Busto, J.V.; Shaw, W.A.; Duan, J.; Merrill, J.A.H.; Alonso, A.; Goñi, F.M. Biophysical Properties of Novel 1-Deoxy-(Dihydro)ceramides Occurring in Mammalian Cells. Biophys. J. 2014, 107, 2850–2859. [Google Scholar] [CrossRef]
- Ando, A.; Oka, M.; Satomi, Y. Deoxysphingolipids and ether-linked diacylglycerols accumulate in the tissues of aged mice. Cell Biosci. 2019, 9, 1–7. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millner, A.; Atilla-Gokcumen, G.E. Lipid Players of Cellular Senescence. Metabolites 2020, 10, 339. https://doi.org/10.3390/metabo10090339
Millner A, Atilla-Gokcumen GE. Lipid Players of Cellular Senescence. Metabolites. 2020; 10(9):339. https://doi.org/10.3390/metabo10090339
Chicago/Turabian StyleMillner, Alec, and G. Ekin Atilla-Gokcumen. 2020. "Lipid Players of Cellular Senescence" Metabolites 10, no. 9: 339. https://doi.org/10.3390/metabo10090339
APA StyleMillner, A., & Atilla-Gokcumen, G. E. (2020). Lipid Players of Cellular Senescence. Metabolites, 10(9), 339. https://doi.org/10.3390/metabo10090339