Machine Learning Applications for Mass Spectrometry-Based Metabolomics

 ,
,  , , and
, , and
Abstract
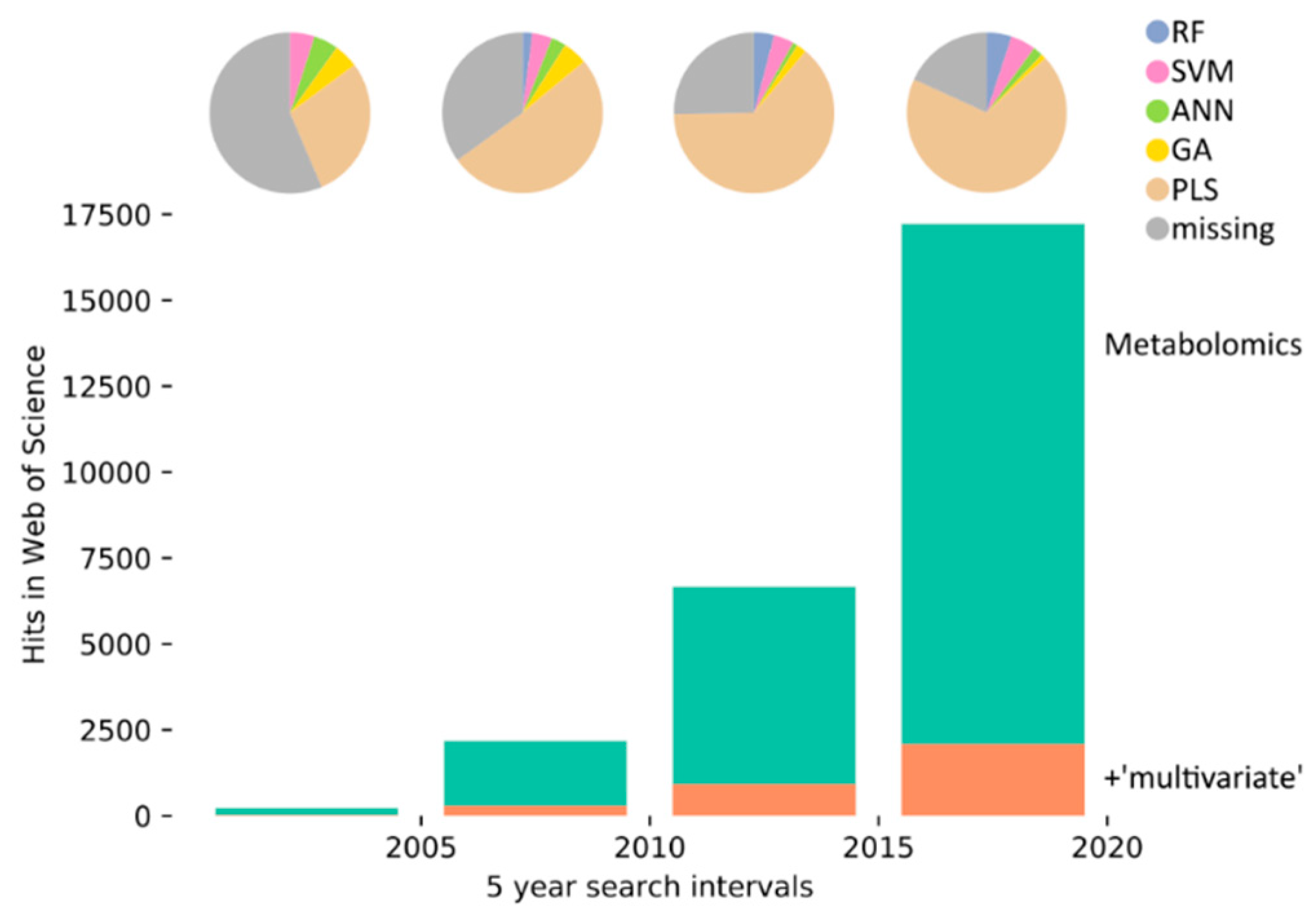
1. Introduction
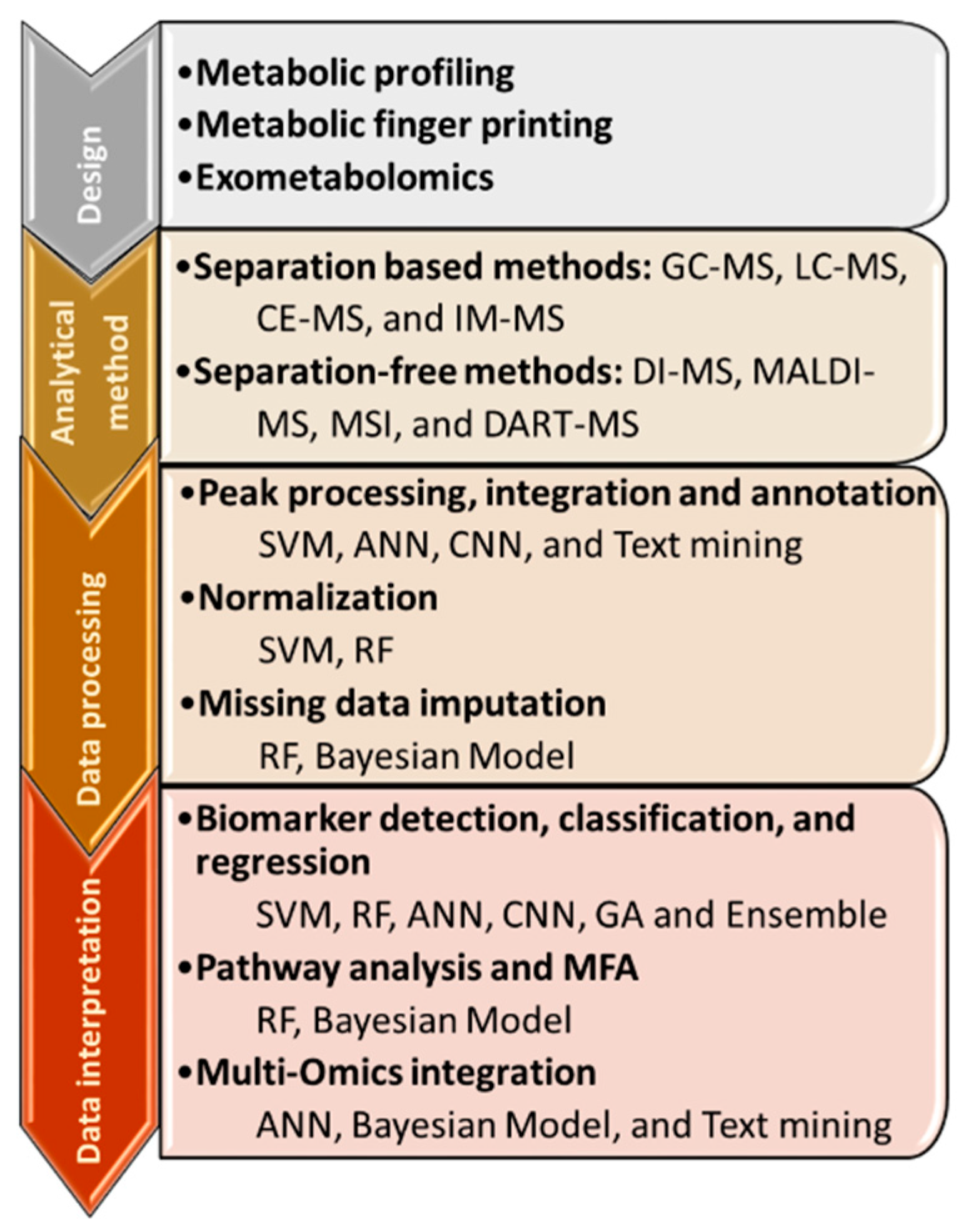
2. Machine Learning for Data Processing
2.1. Peak Picking, Integration and Annotation
2.2. Normalization Procedures
2.3. Missing Data Imputation
3. Biological Insights with Metabolomics
3.1. Biomarker Detection, Classification, and Regression
3.2. Metabolomics to Pathways
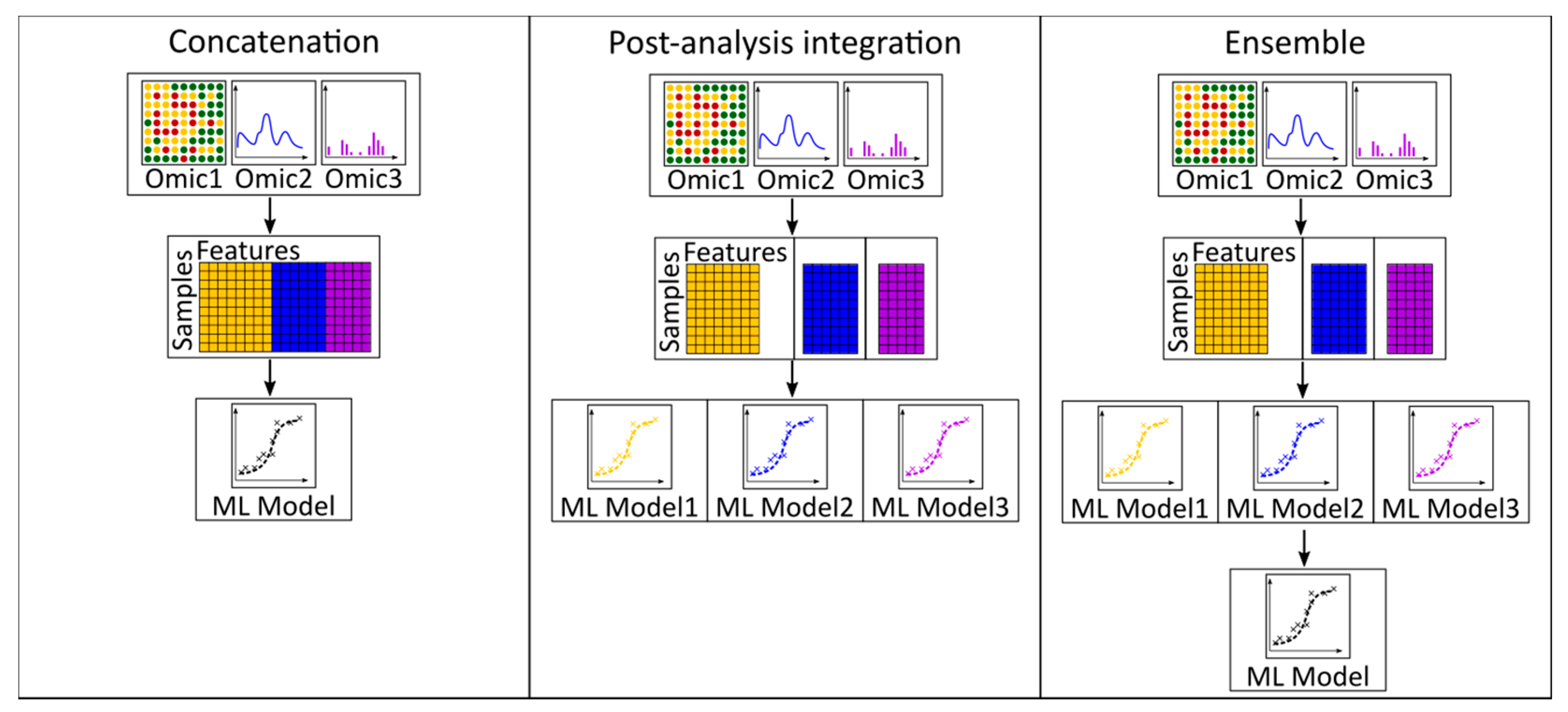
3.3. Multi-Omics Integration
4. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations and Terms
| Activation Function | The function that defines whether a neuron in a neural network is active. |
| Bayesian model | Bayes theorem is used with prior probabilities of past events for prediction. |
| CNN | Convolutional neural networks are a special form of artificial neural networks, strong when feature geometry is important as in images or spectral data. |
| Cross validation | Data is divided into folds, where every fold is used as a test set and average metrics across the folds are used to evaluate model statistics. |
| Feature | Observed variable used as input to the model for prediction. |
| Hyperparameter | Also known as metaparameters and used for tuning of the model training. |
| Latent variables | Features derived by mathematical transformation of features. |
| Overfitting | The model performs well on the training data but poorly on unknown data. Overfitting increases with variables and nonlinearity of the statistical model. Cross validation identifies overfitting. |
References
- Leavell, M.D.; Singh, A.H.; Kaufmann-Malaga, B.B. High-throughput screening for improved microbial cell factories, perspective and promise. Curr. Opin. Biotechnol. 2020, 62, 22–28. [Google Scholar] [CrossRef]
- Saccenti, E.; Hoefsloot, H.C.J.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M.W.B. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- Touw, W.G.; Bayjanov, J.R.; Overmars, L.; Backus, L.; Boekhorst, J.; Wels, M.; van Hijum, S.A.F.T. Data mining in the Life Sciences with Random Forest: A walk in the park or lost in the jungle? Brief. Bioinf. 2013, 14, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Brereton, R.G.; Lloyd, G.R. Support vector machines for classification and regression. Analyst 2010, 135, 230–267. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.J.; Goodacre, R.; Woodward, A.M.; Kell, D.B. Genetic programming: A novel method for the quantitative analysis of pyrolysis mass spectral data. Anal. Chem. 1997, 69, 4381–4389. [Google Scholar] [CrossRef] [PubMed]
- Curry, B.; Rumelhart, D.E. MSnet: A neural network which classifies mass spectra. Tetrahedron Comput. Methodol. 1990, 3, 213–237. [Google Scholar] [CrossRef]
- Cirovic, D.A. Feed-forward artificial neural networks: Applications to spectroscopy. TrAC Trends Anal. Chem. 1997, 16, 148–155. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Mendez, K.M.; Broadhurst, D.I.; Reinke, S.N. The application of artificial neural networks in metabolomics: A historical perspective. Metabolomics 2019, 15, 142. [Google Scholar] [CrossRef]
- Brown, M.P.S.; Grundy, W.N.; Lin, D.; Cristianini, N.; Sugnet, C.W.; Furey, T.S.; Ares, M.; Haussler, D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. USA 2000, 97, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Hong, H.; Fang, H.; Xie, Q.; Perkins, R. Decision forest: Combining the predictions of multiple independent decision tree models. J. Chem. Inf. Comput. Sci. 2003, 43, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Truong, Y.; Lin, X.; Beecher, C. Learning a complex metabolomic dataset using random forests and support vector machines. In Proceedings of the KDD ’04: Proceedings of Knowledge Discovery and Data Mining, Seattle, WA, USA, 22–25 August 2004; pp. 835–840. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Le, T.T.; Fu, W.; Moore, J.H. Scaling tree-based automated machine learning to biomedical big data with a feature set selector. Bioinformatics 2020, 36, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, J. Machine Learning in Untargeted Metabolomics Experiments. Methods Mol. Biol. 2019, 1859, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Liggi, S.; Hinz, C.; Hall, Z.; Santoru, M.L.; Poddighe, S.; Fjeldsted, J.; Atzori, L.; Griffin, J.L. KniMet: A pipeline for the processing of chromatography–mass spectrometry metabolomics data. Metabolomics 2018, 14. [Google Scholar] [CrossRef]
- Chollet, F. Deep Learning with Python; Manning: Shelter Island, NY, USA, 2017. [Google Scholar]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2018, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Aksenov, A.A.; da Silva, R.; Knight, R.; Lopes, N.P.; Dorrestein, P.C. Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem. 2017, 1, 54. [Google Scholar] [CrossRef]
- Blaženović, I.; Kind, T.; Ji, J.; Fiehn, O. Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B.; Mohapatra, S. Tools and resources for metabolomics research community: A 2017–2018 update. Electrophoresis 2018, 40, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, K.; Misra, B.B. Software tools, databases and resources in metabolomics: Updates from 2018 to 2019. Metabolomics 2020, 16, 1–23. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 2012, 30, 826–828. [Google Scholar] [CrossRef]
- Cho, K.; Mahieu, N.; Ivanisevic, J.; Uritboonthai, W.; Chen, Y.J.; Siuzdak, G.; Patti, G.J. isoMETLIN: A database for isotope-based metabolomics. Anal. Chem. 2014, 86, 9358–9361. [Google Scholar] [CrossRef]
- Simón-Manso, Y.; Lowenthal, M.S.; Kilpatrick, L.E.; Sampson, M.L.; Telu, K.H.; Rudnick, P.A.; Mallard, W.G.; Bearden, D.W.; Schock, T.B.; Tchekhovskoi, D.V.; et al. Metabolite Profiling of a NIST Standard Reference Material for Human Plasma (SRM 1950): GC-MS, LC-MS, NMR, and Clinical Laboratory Analyses, Libraries, and Web-Based Resources. Anal. Chem. 2013, 85, 11725–11731. [Google Scholar] [CrossRef] [PubMed]
- Babushok, V.I.; Linstrom, P.J.; Reed, J.J.; Zenkevich, I.G.; Brown, R.L.; Mallard, W.G.; Stein, S.E. Development of a database of gas chromatographic retention properties of organic compounds. J. Chromatogr. A 2007, 1157, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Chikayama, E.; Yuasa, H.; Shimada, Y.; Tohge, T.; Shinozaki, K.; Hirai, M.Y.; Sakurai, T.; Kikuchi, J.; Saito, K. PRIMe: A Web site that assembles tools for metabolomics and transcriptomics. In Silico Biol. 2008, 8, 339–345. [Google Scholar]
- Sakurai, T.; Yamada, Y.; Sawada, Y.; Matsuda, F.; Akiyama, K.; Shinozaki, K.; Hirai, M.Y.; Saito, K. PRIMe Update: Innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol. 2013, 54, e5. [Google Scholar] [CrossRef]
- Hummel, M.; Meister, R.; Mansmann, U. GlobalANCOVA: Exploration and assessment of gene group effects. Bioinformatics 2008, 24, 78–85. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Sawada, Y.; Nakabayashi, R.; Yamada, Y.; Suzuki, M.; Sato, M.; Sakata, A.; Akiyama, K.; Sakurai, T.; Matsuda, F.; Aoki, T.; et al. RIKEN tandem mass spectral database (ReSpect) for phytochemicals: A plant-specific MS/MS-based data resource and database. Phytochemistry 2012, 82, 38–45. [Google Scholar] [CrossRef]
- An, P.N.T.; Fukusaki, E. Metabolomics: State-of-the-Art Technologies and Applications on Drosophila melanogaster. Adv. Exp. Med. Biol. 2018, 1076, 257–276. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Zhang, X.; Wu, C.; Lin, T.; Ying, Y. Deep learning for vibrational spectral analysis: Recent progress and a practical guide. Anal. Chim. Acta 2019, 1081, 6–17. [Google Scholar] [CrossRef]
- Risum, A.B.; Bro, R. Using deep learning to evaluate peaks in chromatographic data. Talanta 2019, 204, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.M.; Lozano, S.; Umbdenstock, T.; Croixmarie, V.; Arrault, A.; Vayer, P. UPLC–MS retention time prediction: A machine learning approach to metabolite identification in untargeted profiling. Metabolomics 2016, 12, 8. [Google Scholar] [CrossRef]
- Creek, D.J.; Jankevics, A.; Breitling, R.; Watson, D.G.; Barrett, M.P.; Burgess, K.E.V. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography–mass spectrometry: Improved metabolite identification by retention time prediction. Anal. Chem. 2011, 83, 8703–8710. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, R.; Martens, L.; Degroeve, S. Comprehensive and empirical evaluation of machine learning algorithms for small molecule LC retention time prediction. Anal. Chem. 2019, 91, 3694–3703. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Guijas, C.; Billings, E.; Montenegro-Burke, J.R.; Uritboonthai, W.; Aisporna, A.E.; Chen, E.; Benton, H.P.; Siuzdak, G. The METLIN small molecule dataset for machine learning-based retention time prediction. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, T.; Xu, J.; Luo, X.; Ying, Y. DeepSpectra: An end-to-end deep learning approach for quantitative spectral analysis. Anal. Chim. Acta 2019, 1058, 48–57. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Nguyen, C.H.; Mamitsuka, H. Recent advances and prospects of computational methods for metabolite identification. Brief. Bioinf. 2019, 20, 2028–2043. [Google Scholar] [CrossRef]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar] [CrossRef]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef]
- Wei, J.N.; Belanger, D.; Adams, R.P.; Sculley, D. Rapid Prediction of Electron–Ionization Mass Spectrometry Using Neural Networks. ACS Cent. Sci. 2019, 5, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Lu, H.; Zhang, Z. Predicting Molecular Fingerprint from Electron–Ionization Mass Spectrum with Deep Neural Networks. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Mrzic, A.; Meysman, P.; De Vijlder, T.; Romijn, E.P.; Valkenborg, D.; Bittremieux, W.; Laukens, K. MESSAR: Automated recommendation of metabolite substructures from tandem mass spectra. PLoS ONE 2020, 15, e0226770. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Ong, C.W.; Wandy, J.; Ernst, M.; Ridder, L.; Van Der Hooft, J.J.J. Deciphering complex metabolite mixtures by unsupervised and supervised substructure discovery and semi-automated annotation from MS/MS spectra. Faraday Discuss 2019, 218, 284–302. [Google Scholar] [CrossRef] [PubMed]
- Borgsmüller, N.; Gloaguen, Y.; Opialla, T.; Blanc, E.; Sicard, E.; Royer, A.-L.; Le Bizec, B.; Durand, S.; Migné, C.; Pétéra, M.; et al. WiPP: Workflow for improved peak picking for gas chromatography-mass spectrometry (GC-MS) data. Metabolites 2019, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Kantz, E.D.; Tiwari, S.; Watrous, J.D.; Cheng, S.; Jain, M. Deep Neural Networks for Classification of LC-MS Spectral Peaks. Anal. Chem. 2019, 91, 12407–12413. [Google Scholar] [CrossRef]
- Liu, Z.; Portero, E.P.; Jian, Y.; Zhao, Y.; Onjiko, R.M.; Zeng, C.; Nemes, P. Trace, Machine Learning of Signal Images for Trace-Sensitive Mass Spectrometry: A Case Study from Single-Cell Metabolomics. Anal. Chem. 2019, 91, 5768–5776. [Google Scholar] [CrossRef]
- Melnikov, A.; Tsentalovich, Y.P.; Yanshole, V.V. Deep learning for the precise peak detection in high-resolution LC-MS data. Anal. Chem. 2019. [Google Scholar] [CrossRef]
- Ji, H.; Lu, H.; Zhang, Z. Deep Learning Enable Untargeted Metabolite Extraction from High Throughput Coverage Data-Independent Acquisition. bioRxiv 2020. [Google Scholar] [CrossRef]
- Seddiki, K.; Saudemont, P.; Precioso, F.; Ogrinc, N.; Wisztorski, M.; Salzet, M.; Fournier, I.; Droit, A. Towards CNN Representations for Small Mass Spectrometry Data Classification: From Transfer Learning to Cumulative Learning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lv, J.; Wei, J.; Wang, Z.; Cao, J. Multiple Compounds Recognition from the Tandem Mass Spectral Data Using Convolutional Neural Network. Molecules 2019, 24, 4590. [Google Scholar] [CrossRef] [PubMed]
- Brouard, C.; Bassé, A.; d’Alché-Buc, F.; Rousu, J. Improved Small Molecule Identification through Learning Combinations of Kernel Regression Models. Metabolites 2019, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Xu, Y.; Lu, H.; Zhang, Z. Deep MS/MS-Aided Structural-similarity Scoring for Unknown Metabolites Identification. Anal. Chem. 2019, 97, 5629–5637. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Kind, T.; Cajka, T.; Hazen, S.L.; Tang, W.H.W.; Kaddurah-Daouk, R.; Irvin, M.R.; Arnett, D.K.; Barupal, D.K.; Fiehn, O. Systematic Error Removal Using Random Forest for Normalizing Large-Scale Untargeted Lipidomics Data. Anal. Chem. 2019, 91, 3590–3596. [Google Scholar] [CrossRef]
- Wang, S.; Yang, H. pseudoQC: A Regression-Based Simulation Software for Correction and Normalization of Complex Metabolomics and Proteomics Datasets. Proteomics 2019, 19, 1900264. [Google Scholar] [CrossRef]
- Vollmar, A.K.R.; Rattray, N.J.W.; Cai, Y.; Santos-Neto, Á.J.; Deziel, N.C.; Jukic, A.M.Z.; Johnson, C.H. Normalizing Untargeted Periconceptional Urinary Metabolomics Data: A Comparison of Approaches. Metabolites 2019, 9, 198. [Google Scholar] [CrossRef]
- Kokla, M.; Virtanen, J.; Kolehmainen, M.; Paananen, J.; Hanhineva, K. Random forest-based imputation outperforms other methods for imputing LC-MS metabolomics data: A comparative study. BMC Bioinf. 2019, 20, 1–11. [Google Scholar] [CrossRef]
- Shah, J.; Brock, G.N.; Gaskins, J. BayesMetab: Treatment of missing values in Metabolomic studies using a Bayesian modeling approach. BMC Bioinf. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Hrydziuszko, O.; Viant, M.R. Missing values in mass spectrometry based metabolomics: An undervalued step in the data processing pipeline. Metabolomics 2012, 8, 161–174. [Google Scholar] [CrossRef]
- Webb-Robertson, B.-J.M.; Wiberg, H.K.; Matzke, M.M.; Brown, J.N.; Wang, J.; McDermott, J.E.; Smith, R.D.; Rodland, K.D.; Metz, T.O.; Pounds, J.G.; et al. Review, Evaluation, and Discussion of the Challenges of Missing Value Imputation for Mass Spectrometry-Based Label-Free Global Proteomics. J. Proteome Res. 2015, 14, 1993–2001. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-based Metabolomics Data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef] [PubMed]
- Di Guida, R.; Engel, J.; Allwood, J.W.; Weber, R.J.M.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B. Non-targeted UHPLC-MS metabolomic data processing methods: A comparative investigation of normalisation, missing value imputation, transformation and scaling. Metabolomics 2016, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, A.; Marwala, T. Evaluating the impact of missing data imputation through the use of the random forest algorithm. arXiv 2008, arXiv:0812.2412. [Google Scholar]
- Lee, M.Y.; Hu, T. Computational methods for the discovery of metabolic markers of complex traits. Metabolites 2019, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Mendez, K.M.; Broadhurst, D.I.; Reinke, S.N. Migrating from partial least squares discriminant analysis to artificial neural networks: A comparison of functionally equivalent visualisation and feature contribution tools using jupyter notebooks. Metabolomics 2020, 16, 17. [Google Scholar] [CrossRef]
- Mendez, K.M.; Reinke, S.N.; Broadhurst, D.I. A comparative evaluation of the generalised predictive ability of eight machine learning algorithms across ten clinical metabolomics data sets for binary classification. Metabolomics 2019, 15, 150. [Google Scholar] [CrossRef]
- Morais, C.L.M.; Lima, K.M.G.; Martin, F.L. Uncertainty estimation and misclassification probability for classification models based on discriminant analysis and support vector machines. Anal. Chim. Acta 2019, 1063, 40–46. [Google Scholar] [CrossRef]
- Perakakis, N.; Polyzos, S.A.; Yazdani, A.; Sala-Vila, A.; Kountouras, J.; Anastasilakis, A.D.; Mantzoros, C.S. Non-invasive diagnosis of non-alcoholic steatohepatitis and fibrosis with the use of omics and supervised learning: A proof of concept study. Metabolism 2019, 101, 154005. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, G.; Sun, M.; Pan, X.; Yang, Z. Integrating a generalized data analysis workflow with the Single-probe mass spectrometry experiment for single cell metabolomics. Anal. Chim. Acta 2019, 1064, 71–79. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, H.; Chen, D.; Zhao, Y.-Y. Machine learning distilled metabolite biomarkers for early stage renal injury. Metabolomics 2020, 16, 4. [Google Scholar] [CrossRef]
- Sirén, K.; Fischer, U.; Vestner, J. Automated supervised learning pipeline for non-targeted GC-MS data analysis. Anal. Chim. Acta X 2019, 1, 100005. [Google Scholar] [CrossRef]
- Peeters, L.; Beirnaert, C.; Van der Auwera, A.; Bijttebier, S.; De Bruyne, T.; Laukens, K.; Pieters, L.; Hermans, N.; Foubert, K. Revelation of the metabolic pathway of Hederacoside C using an innovative data analysis strategy for dynamic multiclass biotransformation experiments. J. Chromatogr. A 2019, 1595, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Reynes, C.; Cazals, G.; Enjalbal, C.; Sabatier, R.; Saucier, C. Discrimination of rosé wines using shotgun metabolomics with a genetic algorithm and MS ion intensity ratios. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Grissa, D.; Comte, B.; Petera, M.; Pujos-Guillot, E.; Napoli, A. A hybrid and exploratory approach to knowledge discovery in metabolomic data. Discret. Appl. Math. 2019. [Google Scholar] [CrossRef]
- Opgenorth, P.; Costello, Z.; Okada, T.; Goyal, G.; Chen, Y.; Gin, J.; Benites, V.T.; Raad, M.d.; Northen, T.R.; Deng, K.; et al. Lessons from two Design-Build-Test-Learn cycles of dodecanol production in Escherichia coli aided by machine learning. ACS Synth. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jahagirdar, S.; Suarez-Diez, M.; Saccenti, E. Simulation and Reconstruction of Metabolite–Metabolite Association Networks Using a Metabolic Dynamic Model and Correlation Based Algorithms. J. Proteome. Res. 2019, 18, 1099–1113. [Google Scholar] [CrossRef]
- Toubiana, D.; Puzis, R.; Wen, L.; Sikron, N.; Kurmanbayeva, A.; Soltabayeva, A.; Wilhelmi, M.D.M.R.; Sade, N.; Fait, A.; Sagi, M.; et al. Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data. Commun. Biol. 2019, 2, 214. [Google Scholar] [CrossRef]
- Hosseini, R.; Hassanpour, N.; Liu, L.-P.; Hassoun, S. Pathway Activity Analysis and Metabolite Annotation for Untargeted Metabolomics using Probabilistic Modeling. arXiv 2019, arXiv:1912.05753. [Google Scholar]
- Machiraju, G.; Amar, D.; Ashley, E. Multi-Omics Factorization Illustrates the Added Value of Deep Learning Approaches; Stanford University: Stanford, CA, USA, 2019. [Google Scholar]
- Le, V.; Quinn, T.P.; Tran, T.; Venkatesh, S. Deep in the Bowel: Highly Interpretable Neural Encoder-Decoder Networks Predict Gut Metabolites from Gut Microbiome. bioRxiv 2019. [Google Scholar] [CrossRef]
- Morton, J.T.; Aksenov, A.A.; Nothias, L.F.; Foulds, J.R.; Quinn, R.A.; Badri, M.H.; Swenson, T.L.; Van Goethem, M.W.; Northen, T.R.; Vazquez-Baeza, Y.; et al. Learning representations of microbe–metabolite interactions. Nat. Methods 2019, 16, 1306–1314. [Google Scholar] [CrossRef]
- John, P.C.S.; Strutz, J.; Broadbelt, L.J.; Tyo, K.E.J.; Bomble, Y.J. Bayesian inference of metabolic kinetics from genome-scale multiomics data. PLoS Comput. Biol. 2019, 15. [Google Scholar] [CrossRef]
- Liebermeister, W. Model balancing: Consistent in-vivo kinetic constants and metabolic states obtained by convex optimisation. bioRxiv 2019. [Google Scholar] [CrossRef]
- Quinn, T.P.; Erb, I. Another look at microbe–metabolite interactions: How scale invariant correlations can outperform a neural network. bioRxiv 2019. [Google Scholar] [CrossRef]
- Morton, J.T.; McDonald, D.; Aksenov, A.A.; Nothias, L.F.; Foulds, J.R.; Quinn, R.A.; Badri, M.H.; Swenson, T.L.; Van Goethem, M.W.; Northen, T.R.; et al. Revisiting microbe-metabolite interactions: Doing better than random. bioRxiv 2019. [Google Scholar] [CrossRef]
- Hira, Z.M.; Gillies, D.F. A Review of Feature Selection and Feature Extraction Methods Applied on Microarray Data. Adv. Bioinform. 2015, 2015. [Google Scholar] [CrossRef]
- Xia, J.; Broadhurst, D.I.; Wilson, M.; Wishart, D.S. Translational biomarker discovery in clinical metabolomics: An introductory tutorial. Metabolomics 2013, 9, 280–299. [Google Scholar] [CrossRef]
- Ali, N.; Girnus, S.; Rösch, P.; Popp, J.; Bocklitz, T. Sample-Size Planning for Multivariate Data: A Raman-Spectroscopy-Based Example. Anal. Chem. 2018, 90, 12485–12492. [Google Scholar] [CrossRef]
- Heinemann, J.; Mazurie, A.; Tokmina-Lukaszewska, M.; Beilman, G.J.; Bothner, B. Application of support vector machines to metabolomics experiments with limited replicates. Metabolomics 2014, 10, 1121–1128. [Google Scholar] [CrossRef]
- van der Ploeg, T.; Austin, P.C.; Steyerberg, E.W. Modern modelling techniques are data hungry: A simulation study for predicting dichotomous endpoints. BMC Med. Res. Methodol. 2014, 14. [Google Scholar] [CrossRef]
- Ivanisevic, J.; Want, E.J. From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data. Metabolites 2019, 9, 308. [Google Scholar] [CrossRef]
- Zampieri, G.; Vijayakumar, S.; Yaneske, E.; Angione, C. Machine and deep learning meet genome-scale metabolic modeling. PLoS Comput. Biol. 2019, 15, e1007084. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.; Berry, C.; Ghosh, P.; Fong, S.S. Recent advances on constraint-based models by integrating machine learning. Curr. Opin. Biotechnol. 2020, 64, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Liebal, U.W.; Millat, T.; de Jong, I.G.; Kuipers, O.P.; Völker, U.; Wolkenhauer, O. How mathematical modelling elucidates signalling in Bacillus subtilis. Mol. Microbiol. 2010, 77, 1083–1095. [Google Scholar] [CrossRef][Green Version]
- Heckmann, D.; Lloyd, C.J.; Mih, N.; Ha, Y.; Zielinski, D.C.; Haiman, Z.B.; Desouki, A.A.; Lercher, M.J.; Palsson, B.O. Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models. Nat. Commun. 2018, 9, 5252. [Google Scholar] [CrossRef] [PubMed]
- Alber, M.; Tepole, A.B.; Cannon, W.R.; De, S.; Dura-Bernal, S.; Garikipati, K.; Karniadakis, G.; Lytton, W.W.; Perdikaris, P.; Petzold, L.; et al. Integrating machine learning and multiscale modeling—Perspectives, challenges, and opportunities in the biological, biomedical, and behavioral sciences. NPJ Digit. Med. 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Costello, Z.; Martin, H.G. A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. NPJ Syst. Biol. Appl. 2018, 4, 19. [Google Scholar] [CrossRef]
- Jervis, A.J.; Carbonell, P.; Vinaixa, M.; Dunstan, M.S.; Hollywood, K.A.; Robinson, C.J.; Rattray, N.J.W.; Yan, C.; Swainston, N.; Currin, A.; et al. Machine Learning of Designed Translational Control Allows Predictive Pathway Optimization in Escherichia coli. ACS Synth. Biol. 2018, 8, 127–136. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, G.; Dong, J.; Xing, X.-H.; Dai, J.; Zhang, C. MiYA, an efficient machine-learning workflow in conjunction with the YeastFab assembly strategy for combinatorial optimization of heterologous metabolic pathways in Saccharomyces cerevisiae. Metab. Eng. 2018, 47, 294–302. [Google Scholar] [CrossRef]
- Karnovsky, A.; Li, S. Pathway Analysis for Targeted and Untargeted Metabolomics. Comput. Methods Data Anal. Metab. 2020, 2104, 367–400. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med. 2018, 50, 19. [Google Scholar] [CrossRef]
- Kogadeeva, M.; Zamboni, N. SUMOFLUX: A Generalized Method for Targeted 13C Metabolic Flux Ratio Analysis. PLoS Comput. Biol. 2016, 12, e1005109. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.G.; Wang, Y.; Jiang, W.; Oyetunde, T.; Yao, R.; Zhang, X.; Shimizu, K.; Tang, Y.J.; Bao, F.S. Rapid prediction of bacterial heterotrophic fluxomics using machine learning and constraint programming. PLoS Comput. Biol. 2016, 12, e1004838. [Google Scholar] [CrossRef] [PubMed]
- Noor, E.; Cherkaoui, S.; Sauer, U. Biological insights through omics data integration. Curr. Opin. Syst. Biol. 2019, 15, 39–47. [Google Scholar] [CrossRef]
- Ritchey, T. General Morphological Analysis (GMA). Wicked Probl. Soc. Messes 2011, 17, 7–18. [Google Scholar] [CrossRef]
- Acharjee, A.; Ament, Z.; West, J.A.; Stanley, E.; Griffin, J.L. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinf. 2016, 17, 440. [Google Scholar] [CrossRef]
- Manor, O.; Zubair, N.; Conomos, M.P.; Xu, X.; Rohwer, J.E.; Krafft, C.E.; Lovejoy, J.C.; Magis, A.T. A Multi-omic Association Study of Trimethylamine N-Oxide. Cell Rep. 2018, 24, 935–946. [Google Scholar] [CrossRef]
- Nam, H.; Chung, B.C.; Kim, Y.; Lee, K.; Lee, D. Combining tissue transcriptomics and urine metabolomics for breast cancer biomarker identification. Bioinformatics 2009, 25, 3151–3157. [Google Scholar] [CrossRef]
- Wang, M.; Yu, G.; Ressom, H.W. Integrative Analysis of Proteomic, Glycomic, and Metabolomic Data for Biomarker Discovery. IEEE J. Biomed. Health Inform. 2016, 20, 1225–1231. [Google Scholar] [CrossRef]
- Miskovic, L.; Beal, J.; Moret, M.; Hatzimanikatis, V. Uncertainty Reduction in Biochemical Kinetic Models: Enforcing Desired Model Properties. PLoS Comput. Biol. 2019, 15, e1007242. [Google Scholar] [CrossRef]
- Andreozzi, S.; Miskovic, L.; Hatzimanikatis, V. iSCHRUNK–in silico approach to characterization and reduction of uncertainty in the kinetic models of genome-scale metabolic networks. Metab. Eng. 2016, 33, 158–168. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—A framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Shannon, C.P.; Gautier, B.T.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Lê Cao, K.-A. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Bergdahl, B.; Machado, D.; Dato, L.; Han, T.-L.; Li, J.; Villas-Boas, S.; Herrgård, M.J.; Förster, J.; Panagiotou, G. Linking genetic, metabolic, and phenotypic diversity among Saccharomyces cerevisiae strains using multi-omics associations. GigaScience 2019, 8, giz015. [Google Scholar] [CrossRef]
- Pirhaji, L.; Milani, P.; Leidl, M.; Curran, T.; Avila-Pacheco, J.; Clish, C.B.; White, F.M.; Saghatelian, A.; Fraenkel, E. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 2016, 13, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Zhou, Y.; Ressom, H.W. MOTA: Network-Based Multi-Omic Data Integration for Biomarker Discovery. Metabolites 2020, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Hernandez Gardiol, D.; Chiappino Pepe, A.; Hatzimanikatis, V. TEX-FBA: A constraint-based method for integrating gene expression, thermodynamics, and metabolomics data into genome-scale metabolic models. bioRxiv 2019. [Google Scholar] [CrossRef]
- Hadadi, N.; Pandey, V.; Chiappino-Pepe, A.; Morales, M.; Gallart-Ayala, H.; Mehl, F.; Ivanisevic, J.; Sentchilo, V.; van der Meer, J.R. Mechanistic insights into bacterial metabolic reprogramming from omics-integrated genome-scale models. NPJ Syst. Biol. Appl. 2020, 6, 1–11. [Google Scholar] [CrossRef]
- Huang, M.N.; Yu, W.; Teoh, W.W.; Ardin, M.; Jusakul, A.; Ng, A.W.T.; Boot, A.; Abedi-Ardekani, B.; Villar, S.; Myint, S.S.; et al. Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors. Genome Res. 2017, 27, 1475–1486. [Google Scholar] [CrossRef]
- Zelezniak, A.; Vowinckel, J.; Capuano, F.; Messner, C.B.; Demichev, V.; Polowsky, N.; Mülleder, M.; Kamrad, S.; Klaus, B.; Keller, M.A.; et al. Machine Learning Predicts the Yeast Metabolome from the Quantitative Proteome of Kinase Knockouts. Cell Syst. 2018, 7, 269–283. [Google Scholar] [CrossRef]
- Huang, S.S.Y.; Benskin, J.P.; Veldhoen, N.; Chandramouli, B.; Butler, H.; Helbing, C.C.; Cosgrove, J.R. A multi-omic approach to elucidate low-dose effects of xenobiotics in zebrafish (Danio rerio ) larvae. Aquat. Toxicol. 2017, 182, 102–112. [Google Scholar] [CrossRef]
- Kim, M.; Rai, N.; Zorraquino, V.; Tagkopoulos, I. Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; George, K.W.; Alonso-Gutierrez, J.; Thompson, M.; Baidoo, E.; Wang, G.; Petzold, C.J.; McCloskey, D.; Monk, J.; Yang, L.; et al. Characterizing strain variation in engineered E. coli using a multi-omics-based workflow. Cell Syst. 2016, 2, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Dugourd, A.; Kuppe, C.; Sciacovelli, M.; Gjerga, E.; Emdal, K.B.; Bekker-Jensen, D.B.; Kranz, J.; Bindels, E.J.M.; Costa, S.; Olsen, J.V.; et al. Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dahlin, J.; Holkenbrink, C.; Marella, E.R.; Wang, G.; Liebal, U.; Lieven, C.; Weber, D.; McCloskey, D.; Ebert, B.E.; Herrgard, M.J.; et al. Multi-omics analysis of fatty alcohol production in engineered yeasts Saccharomyces cerevisiae and Yarrowia lipolytica. Front. Genet. 2019, 10, 747. [Google Scholar] [CrossRef]
- Wolpert, D.H.; Macready, W.G. No free lunch theorems for optimization. IEEE Trans. Evol. Comput. 1997, 1, 67–82. [Google Scholar] [CrossRef]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Supervised ML Model | Advantage | Disadvantage | |
|---|---|---|---|
| PLS—Projection to Latent Structure/Partial Linear Regression [2] | |||
 | PLS is a supervised method to construct predictive models when the factors are collinear. PLS-DA is an extension of PLS that can maximize the covariance between classes. Orthogonal PLS (OPLS) is an extension to increase latent feature interpretability. | Overfitting risk: Low Interpretation: High Features/sample: High Implementation: Easy | Collinear data |
| RF—Random Forest [3] | |||
 | Composed of several decision trees. Each decision tree separates the samples according to the measured feature properties. Different trees use a random subset of samples and features for classification. | Overfitting risk: Medium Interpretation: High Implementation: Easy | Features/sample: Low |
| SVM—Support Vector Machine [4] | |||
 | A boundary is determined that separates the classes. For nonlinear separation, the data is augmented by additional dimensions using a kernel function (Φ), often the Radial Basis Function (RBF). | Features/sample: High Implementation: Easy | Overfitting risk: High Interpretation: low |
| ANN—Artificial Neural Network [5] | |||
 | The features are transformed by hidden nodes with a linear equation ‘z’ and a nonlinear function ‘g.’ Several layers may follow, each with nodes containing transformations by functions ‘z’ and ‘g.’ The output is generated by a ‘softmax’ function. | Features/sample: Medium | Overfitting risk: High Interpretation: Medium Implementation: Complex |
| GA—Genetic Algorithm [6] | |||
 | Solution space is searched by operations similar to natural genetic processes to identify suitable solutions. Fitness function is defined to find the fittest solutions. The fittest solutions are subject to cross-over and mutations to evolve towards the best solution. | Interpretation: High | Overfitting risk: High Features/sample: Medium Implementation: Complex |
| Database | Description | URL |
|---|---|---|
| HMDB [29] | 114,193 metabolite entries including both polar and non-polar metabolites | https://hmdb.ca |
| LMSD [30] | 43,665 lipid structures with MS/MS spectra | www.lipidmaps.org/data/structure |
| METLIN [31] | 961,829 molecules (lipids, steroids, plant and bacteria metabolites, small peptides, carbohydrates, exogenous drugs/metabolites, central carbon metabolites and toxicants). Over 14,000 metabolites have been individually analyzed and another 200,000 have in silico MS/MS data | http://metlin.scripps.edu |
| isoMETLIN [32] | All computed isotopologues derived from METLIN based on m/z values and specified isotopes of interest (13C or 15N) | http://isometlin.scripps.edu |
| NIST [33,34] | Reference mass spectra for GC/MS, LC–MS/MS, NMR and gas-phase retention indices for GC | https://chemdata.nist.gov |
| MassBank [35] | Shared public repository of mass spectral data with 41,092 spectra | https://massbank.eu/MassBank |
| MoNA | 200,000+ mass spectral records from experimental, in silico libraries and user contributions | https://mona.fiehnlab.ucdavis.edu |
| mzCloud | More than 6 million multi-stage MSn spectra for more than 17,670 compounds | https://www.mzcloud.org |
| PRIME [36,37] | Standard spectrum of standard compounds generated by GC/MS, LC–MS, CE/MS and NMR | http://prime.psc.riken.jp/ |
| Golm metabolome [38] | 2019 metabolites with GC-MS spectra and retention time indices | http://gmd.mpimp-golm.mpg.de |
| GNPS [39] | Community database for natural products | https://gnps.ucsd.edu |
| ReSpect [40] | Over 9000 MS/MS spectrum of phytochemicals | http://spectra.psc.riken.jp |
| Step | ML Tool | Example | Ref. |
|---|---|---|---|
| Peak picking/integration | SVM | WIPP software: optimization of peak detection, instrument and sample specific | [57] |
| ANN | Peak quality selection for downstream analysis | [58] | |
| CNN | Trace: two-dimensional peak picking over retention time and m/z | [59] | |
| CNN | peakonly software: peak picking and integration | [60] | |
| CNN | Peak classification for subsequent PARAFAC analysis | [43] | |
| CNN | DeepSWATH software: correlation between parent metabolites and fragment ions in MS/MS spectra | [61] | |
| CNN | Representational learning from different tissues, organisms, ionization, instruments for improved classification on small datasets | [62] | |
| CNN | ‘DeepSpectra’: targeted metabolomics on environmental samples, raw spectra analysis | [49] | |
| CNN | Compound recognition in complex tandem MS data tested with several ML tools | [63] | |
| Retention time prediction | ANN | Metlin-integrated prediction of metabolite retention time extrapolation to different chromatographic methods | [48] |
| Ensemble | Performance test of multiple ML algorithms for retention time prediction based on physical properties, ANN and SVM perform well, ensemble training is optimal | [47] | |
| Metabolite annotation | SVM | Input–output kernel regression (IOKR) to predict fingerprint vectors from m/z spectra, mapping molecular structures to spectra | [64] |
| SVM | CSI:Fingerprint:Structure mapping | [52] | |
| Text mining | MS2LDA software: detection of peak co-occurrence | [56] | |
| Text mining | MESSAR software: automated substructure recommendation for co-occurring peaks | [55] | |
| ANN | NEIMS software: ‘neural electron-ionization MS’ spectrum prediction | [54] | |
| ANN | DeepMASS software: substructure detection by comparing unknown spectra to known spectra | [65] | |
| CNN | DeepEI software: fingerprint prediction from MS spectrum | [54] | |
| Normalization | RF | SERRF software: Systematic error removal based on quality control pool samples | [66] |
| RF | pseudoQC software: simulated quality control sample generation, preferably with RF | [67] | |
| SVM | Improvement of statistical analysis by SVM normalization | [68] | |
| Imputation | RF | Best overall performance of RF for unknown missing value type | [69] |
| Bayesian Model | BayesMetab: classification of missing value type, Markov chain Monte Carlo approach with data augmentation | [70] |
| Biological Insight | Optimal ML | Other Models | Samples | Dimension Reduction | Spec-Type | Comment | Ref. |
|---|---|---|---|---|---|---|---|
| Class + biomarker | SVM | LDA, QDA | 4 DS: 30, 280, 240, 183 | PCA | IR | Effect of variance and covariance on classification of infrared spectra. | [79] |
| SVM | RF, PLS-DA | 80 | RFE | LC–MS | Serum identification of lipids, glycans, fatty acids. | [80] | |
| RF | N.A. | <100 | N.A. | SCMS | Single-cell MS on drug response, pathway inference. | [81] | |
| RF | SVM, ANN, CNN | 703 | LASSO | LC–MS | Serum metabolomics related to chronic kidney disease. | [82] | |
| RF | N.A. | 3 DS: 39, 160, 79 | Peak-binning | GCMS | Chromatogram peak ranking for sample discrimination. | [83] | |
| RF | N.A. | 217 | Human selection | LC–MS | Metabolite selection based on expert classification with tinderest Shiny-App. | [84] | |
| ANN | PLS-DA, RF SVM | 10 DS: 968, 253, 668, 59, 184, 97, 80, 100, 121, 83 | N.A. | Bench. data | Thorough comparison of ML approaches on different published targeted MS datasets. | [78] | |
| GA | RF | 60 | N.A. | LC–MS | Wine origin classification. | [85] | |
| Ens. | RF, SVM | 111 | Correlation, information filter | N.A. | Use of symbolic methods, analysis of spectrogram. | [86] | |
| Regression | Ens. | RF, ANN | 2 DS: 36, 60 | N.A. | Assay | Optimization of gene expression for metabolite overproduction. | [87] |
| Pathway inference | RF | Bayes | 500 | N.A. | Sim. | Metabolite correlation network on simulated data. | [88] |
| RF | PLS, Bayes | 339 | Information filter | GCMS | Mapping of metabolic correlation networks to metabolic pathways. | [89] | |
| Bayes | N.A. | 2 DS: 8711, 384 | N.A. | Sim. | ‘PUMA’: Probabilistic modeling for Untargeted Metabolomics Analysis. Simulation of pathway activity, metabolite association, and spectra. | [90] | |
| Multi-omics integration | ANN | SVM | 2 DS: 600, >10,000 | Encoder-decoder | LC–MS/MS | Multi-omics projection to 20–70 latent variables. Classification of latent variables. | [91] |
| ANN | N.A. | 2 DS: 191 in: 1692 out, 51 in: 143 out | Encoder-decoder | LC–MS | Correlation of gut bacteria level to metabolite level, unsupervised clustering of latent variables. | [92] | |
| Text Mining | N.A. | 4 DS: 138 in: 462 out, 466 in: 85 out, 902 in: >10k out, 562 in: > 10k out | N.A. | Bench. data | ‘mmvec’: microbial sequence to metabolite occurrence mapping with as little as 166 microbes mapped to 85 metabolites | [93] | |
| Bayes | N.A. | 25 | N.A | Sim. | Estimation of metabolic kinetics based on multi-omics data for lysine synthesis. | [94] | |
| Bayes | N.A. | 22 | N.A. | Estimation of metabolic kinetics based on multi-omics data | [95] |
| Type | Method | Description | Advantages | Disadvantages |
|---|---|---|---|---|
| Unsupervised method | ||||
| FE | Principal Component Analysis (PCA) | Unsupervised method to transform data into axes that explain maximum variability. Returns orthogonal features. | Prior Information: None | Interpretation: Low |
| FE | Kernel PCA (k-PCA) | Transforms the data into a lower dimension that is linearly separable. | Correlation type: Nonlinear data | Interpretation: Low |
| FE | Encoder–Decoder | ANN-based, the encoder maps input to lower-dimensional latent variables. The decoder uses latent variables to generate output. | Correlation type: Nonlinear data Prior Information: None | Correlation type: Fails on independent data |
| Regularization | ||||
| FS | LASSO or L1 | Supervised method to select sparse features. Regularization parameter (L1 penalty) can be used for regression and classification problems. The coefficients (w) of the features (m) are directly multiplied with the regularization parameter (λ). L1: λ | Interpretation: High | Correlation type: Linear data Note: Minimum selection of features equal to sample size |
| FS | Ridge or L2 | Supervised method to penalize (L2 penalty) large individual weights. The coefficients (w) of the features (m) are squared and multiplied with the regularization parameter (λ). L2: λ | Note: Avoids overfitting | Note: Features are not removed, weights indicate feature importance |
| FS | Elastic Net | Regularization method to retain advantages of both L1 and L2 penalty. EN: λ1 λ2 | Note: Removes features without overfitting | Correlation type: Linear data |
| Discriminant Analysis | ||||
| FE | Linear Discriminant Analysis (LDA) | Supervised method to transform data into axes, which maximizes class separation. Assumes that data is normal with common class covariance. | Prior information: Class labels | Correlation type: Linear data Interpretation: Low |
| Quadratic Discriminant Analysis (QDA) | Supervised classification similar to LDA. Assumes that data is normal but allows for differing class covariance. | Correlation type: Squared nonlinear data | Not useful for dimensionality reduction | |
| Sequential Feature Selection | ||||
| FS | Recursive Feature Elimination/Sequential Backward Selection | At each step, the feature with minimal contribution to the model is dropped until required number of features remain. | Interpretation: High | Note: Optimum not guaranteed |
| Data | Integration Method | Dimensionality Reduction | Model Organisms | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MT | MP | MTP | MPF | MTPF | Concatenation | Post-Analysis Integration | Ensemble | PCA | Regularization | LDA | SFE | Escherichia coli | Danio rerio | Saccharomyces cerevisiae | Mammalian | ||
| Model | Partial least squares | [131] | [109] | [132] | [131,133] | [132] | [133] | [132] | [131] | ||||||||
| Random forest | [120] | [119] | [132] | [122] | [119] | [118,120] | [132] | [118] | [132] | [118,119,120] | |||||||
| SVM | [120,121] | [132] | [121] | [120] | [132] | [121] | [132] | [120,121] | |||||||||
| Artificial neural network | [132] | [134] | [134] | [132,134] | [134] | [132] | |||||||||||
| Genetic algorithms | [109] | [109] | |||||||||||||||
| Bayesian models | [94,95,122] | [95] | [95] | [94,122] | |||||||||||||
| Data | MT | [121,131,133] | [120] | [120] | [121] | [131] | [120,121] | ||||||||||
| MP | [119] | [135] | [135] | [109,135] | [119] | ||||||||||||
| MTP | [132] | [132] | [136] | ||||||||||||||
| MPF | [122] | [94,122] | |||||||||||||||
| MTPF | [137] | [134] | [137] | [134] | [134] | ||||||||||||
| Integration Method | Concatenation | [121] | [131] | [119,121] | |||||||||||||
| Post-analysis integration | [135,137] | [120] | [118] | [135] | [118,120] | ||||||||||||
| Ensemble | [134] | [134] | |||||||||||||||
| Dimensionality reduction | PCA | [135] | [137] | ||||||||||||||
| Regularization | [134] | [132] | |||||||||||||||
| LDA | [120] | ||||||||||||||||
| SFE | [118,121] | ||||||||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243. https://doi.org/10.3390/metabo10060243
Liebal UW, Phan ANT, Sudhakar M, Raman K, Blank LM. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites. 2020; 10(6):243. https://doi.org/10.3390/metabo10060243
Chicago/Turabian StyleLiebal, Ulf W., An N. T. Phan, Malvika Sudhakar, Karthik Raman, and Lars M. Blank. 2020. "Machine Learning Applications for Mass Spectrometry-Based Metabolomics" Metabolites 10, no. 6: 243. https://doi.org/10.3390/metabo10060243
APA StyleLiebal, U. W., Phan, A. N. T., Sudhakar, M., Raman, K., & Blank, L. M. (2020). Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites, 10(6), 243. https://doi.org/10.3390/metabo10060243