Antibiotic-Induced Changes in Microbiome-Related Metabolites and Bile Acids in Rat Plasma

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Clinical Signs
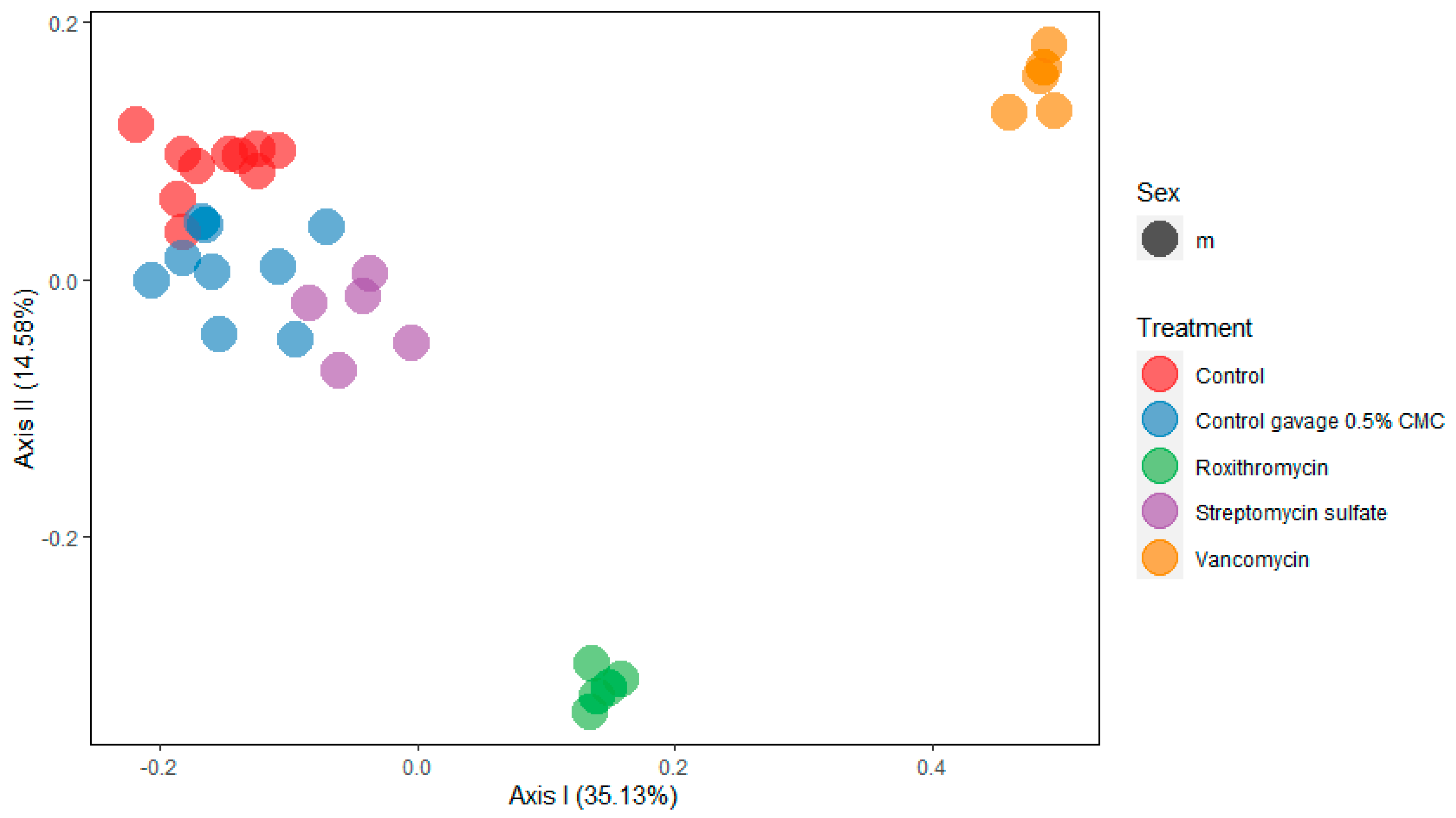
2.2. 16S rRNA Community Analysis
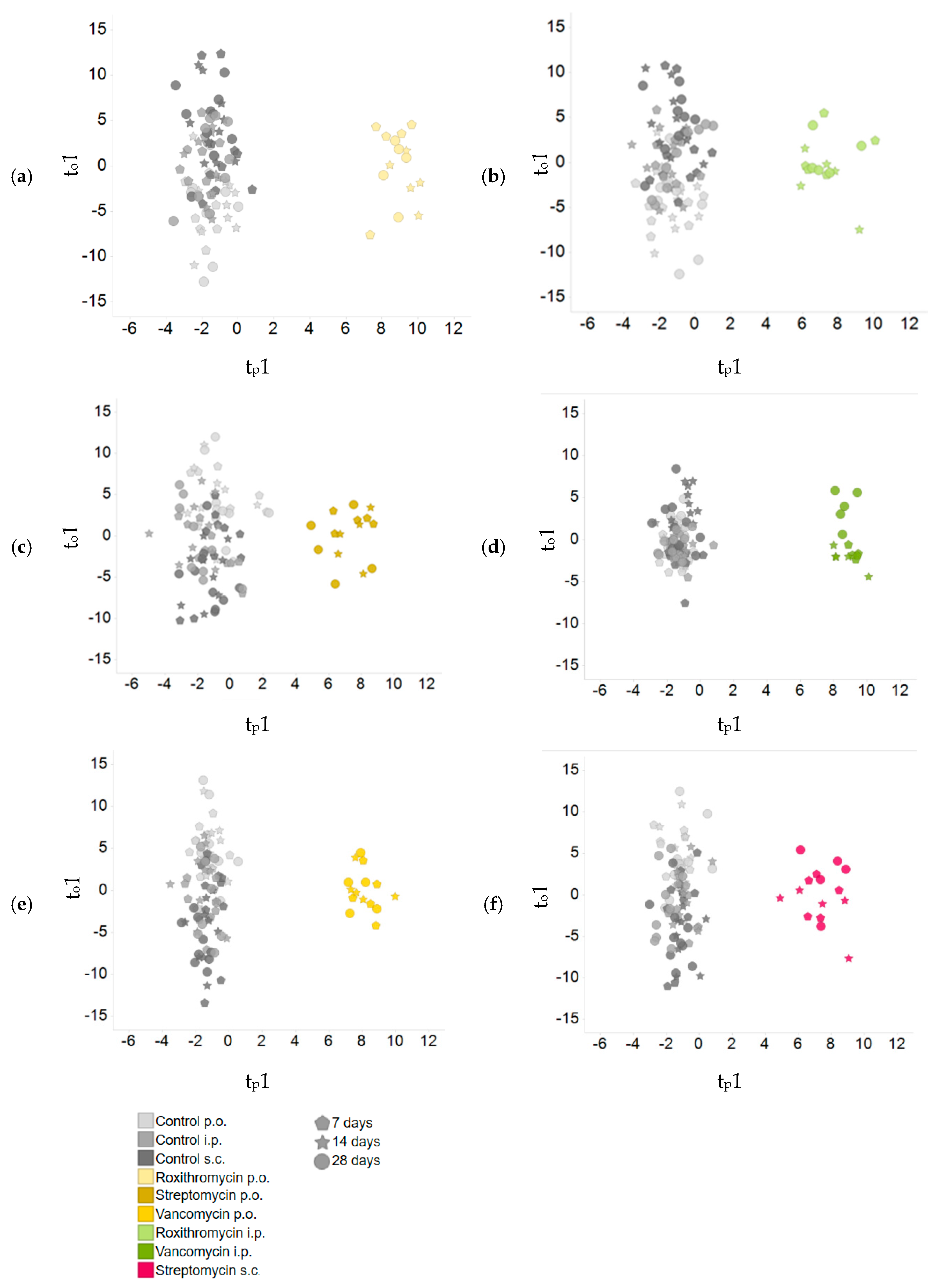
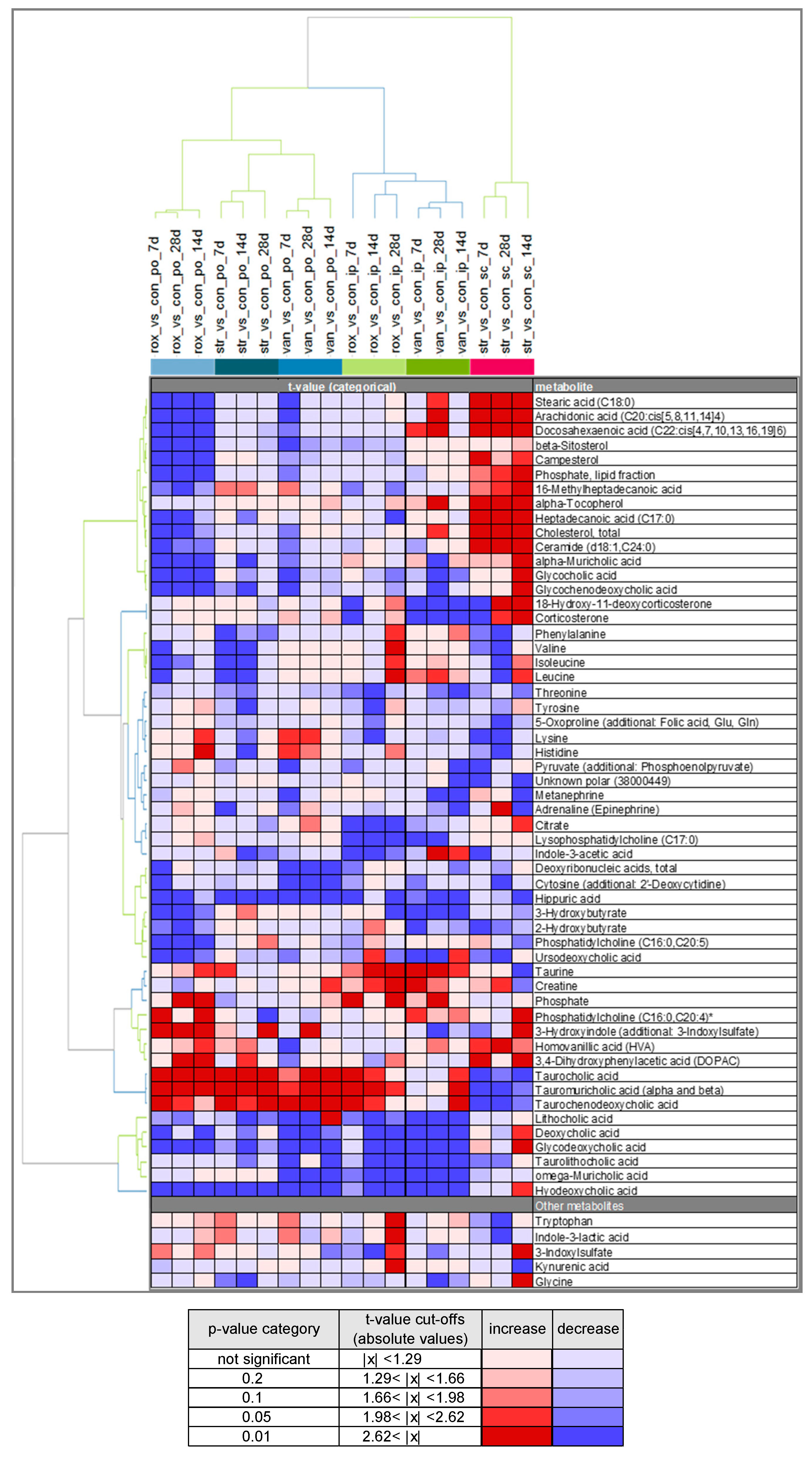
2.3. Comparison of Plasma Metabolite Patterns after Oral and Parenteral Antibiotic-Administration
2.4. Variability in Plasma Bile Acid Levels
3. Discussion
4. Materials and Methods
4.1. Treatment of the Animals
4.2. Ethics Statement
4.3. Blood Sampling
4.4. Clinical Examinations
4.5. Metabolite Profiling
Broad Profiling
4.6. Targeted Bile Acid Analysis
4.7. Community Analysis
4.8. Bioinformatics
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAALAC | Association for Assessment and Accreditation of Laboratory Animal Care International |
| CKD | chronic kidney disease |
| CMC | carboxymethylcellulose |
| Da | Dalton |
| ESI | electrospray ionisation |
| FGF | fibroblast growth factor |
| FXR | farnesoid X receptor |
| GC | gas chromatography |
| GCDCA | glycochenodeoxycholic acid |
| HA | hippuric acid |
| HCA | hierarchical clustering analysis |
| i.p. | intraperitoneal |
| IAA | indole-3-acetic acid |
| IS | 3-indoxylsulfate |
| ID | inner diameter |
| LC | liquid chromatography |
| MS | mass spectrometry |
| MRM | multiple reaction monitoring |
| MTBE | 2-Methoxy-2-methylpropan |
| OECD | Organisation for Economic Co-operation and Development |
| O-PLS-DA | orthogonal projection of latent structures |
| p.o. | per os, by mouth |
| PCA | principal component analysis |
| PCoA | principal coordinate analysis |
| PCR | Polymerase chain reaction |
| s.c. | subcutaneous |
| SPE | solid phase extraction |
| TCA | taurocholic acid |
| TCDCA | taurochenodeoxycholic acid |
| TGR5 | trans-membrane-bound G-protein-coupled receptor |
| UHPLC-ESI | ultra-high-performance liquid chromatography-electrospray ionization |
| VDR | vitamin D receptor |
| QIIME | Quantitative Insights Into Microbial Ecology |
| α-MCA | alpha-muricholic acid |
References
- Zhang, Y.-J.; Li, S.; Gan, R.; Zhou, T.; Xu, N.-P.; Li, H.-B. Impacts of Gut Bacteria on Human Health and Diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef] [PubMed]
- Behr, C.; Kamp, H.; Fabian, E.; Krennrich, G.; Mellert, W.; Peter, E.; Strauss, V.; Walk, T.; Rietjens, I.M.C.M.; Van Ravenzwaay, B. Gut microbiome-related metabolic changes in plasma of antibiotic-treated rats. Arch. Toxicol. 2017, 91, 3439–3454. [Google Scholar] [CrossRef] [PubMed]
- Lindon, J.; Holmes, E.; Bollard, M.E.; Stanley, E.G.; Nicholson, J. Metabonomics technologies and their applications in physiological monitoring, drug safety assessment and disease diagnosis. Biomarkers 2004, 9, 1–31. [Google Scholar] [CrossRef]
- Ramirez, T.; Daneshian, M.; Kamp, H.; Bois, F.; Clench, M.R.; Coen, M.; Donley, B.; Fischer, S.M.; Ekman, E.R.; Fabian, E.; et al. Metabolomics in Toxicology and Preclinical Research. ALTEX 2013, 30, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570–580. [Google Scholar] [CrossRef]
- Begley, M.; Hill, C.; Gahan, C.G. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017, 1, 3–9. [Google Scholar] [CrossRef]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2005, 47, 241–259. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2016, 101, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2010, 60, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; De Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying Diet-Induced Metabolic Changes of the Human Gut Microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; De Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505–4510. [Google Scholar] [CrossRef]
- Brown, S.A.; Riviere, J.E. Comparative pharmacokinetics of aminoglycoside antibiotics. J. Vet. Pharmacol. Ther. 1991, 14, 1–35. [Google Scholar] [CrossRef]
- Musther, H.; Olivares-Morales, A.; Hatley, O.J.; Liu, B.; Rostami-Hodjegan, A. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 2013, 57, 280–291. [Google Scholar] [CrossRef]
- Geraci, J.; Heilman, F.; Nichols, D.; Wellman, W.; Ross, G.; Dorothy, R. Some laboratory and clinical experiences with a new antibiotic, vancomycin. Proc. Staff Meet. Mayo Clin. 1956, 31, 564–582. [Google Scholar]
- Arimori, K.; Miyamoto, S.; Fukuda, K.; Nakamura, C.; Nakano, M. Characteristic difference in gastrointestinal excretion of clarithromycin and roxithromycin. Biopharm. Drug Dispos. 1998, 19, 433–438. [Google Scholar] [CrossRef]
- Currie, B.P.; Lemos-Filho, L. Evidence for Biliary Excretion of Vancomycin into Stool during Intravenous Therapy: Potential Implications for Rectal Colonization with Vancomycin-Resistant Enterococci. Antimicrob. Agents Chemother. 2004, 48, 4427–4429. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Lichtman, J.S.; Ferreyra, J.A.; Ng, K.M.; Smits, S.A.; Sonnenburg, J.L.; Elias, J.E. Host-Microbiota Interactions in the Pathogenesis of Antibiotic-Associated Diseases. Cell Rep. 2016, 14, 1049–1061. [Google Scholar] [CrossRef]
- Preidis, G.; Versalovic, J. Targeting the Human Microbiome with Antibiotics, Probiotics, and Prebiotics: Gastroenterology Enters the Metagenomics Era. Gastroenterology 2009, 136, 2015–2031. [Google Scholar] [CrossRef]
- Hungin, A.P.S.; Mitchell, C.R.; Whorwell, P.; Mulligan, C.; Cole, O.; Agréus, L.; Fracasso, P.; Lionis, C.; Mendive, J.; De Foy, J.-M.P.; et al. Systematic review: Probiotics in the management of lower gastrointestinal symptoms—An updated evidence-based international consensus. Aliment. Pharmacol. Ther. 2018, 47, 1054–1070. [Google Scholar] [CrossRef]
- Herrmann, K.M. The shikimate pathway: Early steps in the biosynthesis of aromatic compounds. Plant Cell 1995, 7, 907. [Google Scholar] [CrossRef]
- Paley, E.L. Diet-Related Metabolic Perturbations of Gut Microbial Shikimate Pathway-Tryptamine-tRNA Aminoacylation-Protein Synthesis in Human Health and Disease. Int. J. Tryptophan Res. 2019, 12. [Google Scholar] [CrossRef]
- Gertsman, I.; Gangoiti, J.A.; Nyhan, W.L.; Barshop, B. Perturbations of tyrosine metabolism promote the indolepyruvate pathway via tryptophan in host and microbiome. Mol. Genet. Metab. 2015, 114, 431–437. [Google Scholar] [CrossRef]
- Devlin, A.S.; Marcobal, A.; Dodd, D.; Nayfach, S.; Plummer, N.; Meyer, T.; Pollard, K.S.; Sonnenburg, J.L.; Fischbach, M.A. Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota. Cell Host Microbe 2016, 20, 709–715. [Google Scholar] [CrossRef]
- Botsford, J.L.; Demoss, R.D. Escherichia coli Tryptophanase in the Enteric Environment. J. Bacteriol. 1972, 109, 74–80. [Google Scholar] [CrossRef]
- Lees, H.J.; Swann, J.R.; Wilson, I.D.; Nicholson, J.; Holmes, E. Hippurate: The Natural History of a Mammalian–Microbial Cometabolite. J. Proteome Res. 2013, 12, 1527–1546. [Google Scholar] [CrossRef]
- Poon, K.; Pang, K.S. Benzoic acid glycine conjugation in the isolated perfused rat kidney. Drug Metab. Dispos. 1995, 23. [Google Scholar]
- Strahl, N.R.; Barr, W.H. Intestinal Drug Absorption and Metabolism III: Glycine Conjugation and Accumulation of Benzoic Acid in Rat Intestinal Tissue. J. Pharm. Sci. 1971, 60, 278–281. [Google Scholar] [CrossRef]
- Calvani, R.; Capuani, G.; Miccheli, A.T.; Puccetti, C.; Delfini, M.; Iaconelli, A.; Nanni, G.; Mingrone, G. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int. J. Obes. 2010, 34, 1095–1098. [Google Scholar] [CrossRef]
- Williams, R.; Eyton-Jones, H.W.; Farnworth, M.J.; Gallagher, R.; Provan, W.M. Effect of intestinal microflora on the urinary metabolic profile of rats: A 1 H-nuclear magnetic resonance spectroscopy study. Xenobiotica 2002, 32, 783–794. [Google Scholar] [CrossRef]
- Zheng, X.; Xie, G.; Zhao, A.; Zhao, L.; Yao, C.; Chiu, N.H.L.; Zhou, Z.; Bao, Y.; Jia, W.; Nicholson, J.; et al. The Footprints of Gut Microbial–Mammalian Co-Metabolism. J. Proteome Res. 2011, 10, 5512–5522. [Google Scholar] [CrossRef]
- Marsot, A.; Boulamery, A.; Bruguerolle, B.; Simon, N. Vancomycin. Clin. Pharmacokinet. 2012, 51, 1–13. [Google Scholar] [CrossRef]
- Motta, I.; Calcagno, A.; Bonora, S. Pharmacokinetics and pharmacogenetics of anti-tubercular drugs: A tool for treatment optimization? Expert Opin. Drug Metab. Toxicol. 2017, 14, 59–82. [Google Scholar] [CrossRef]
- Ryu, S.H.; Kim, J.W.; Yoon, D.; Kim, S.; Kim, K.-B. Serum and urine toxicometabolomics following gentamicin-induced nephrotoxicity in male Sprague-Dawley rats. J. Toxicol. Environ. Heal. Part A 2018, 81, 408–420. [Google Scholar] [CrossRef]
- Harrison, P.M.; Stewart, G.T. Excretion of Antibiotics in Bile. Br. J. Pharmacol. Chemother. 1961, 17, 420–423. [Google Scholar] [CrossRef]
- Robinson, H.J. Streptomycin and Streptothricin: The Absorption, Excretion, and Chemotherapeutic Properties. Ann. N. Y. Acad. Sci. 1946, 48, 119–142. [Google Scholar] [CrossRef]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the gut microbiome in uremia: A potential therapeutic target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The Cardiovascular Effect of the Uremic Solute Indole-3 Acetic Acid. J. Am. Soc. Nephrol. 2014, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Ohtsuki, S.; Otagiri, M.; Takanaga, H.; Asaba, H.; Mori, S.; Terasaki, T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002, 61, 1760–1768. [Google Scholar] [CrossRef]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2010, 108, 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Noshiro, M.; Nishimoto, M.; Okuda, K. Rat liver cholesterol 7 alpha-hydroxylase. Pretranslational regulation for circadian rhythm. J. Boil. Chem. 1990, 265, 10036–10041. [Google Scholar]
- Nakano, A.; Tietz, P.S.; LaRusso, N.F. Circadian rhythms of biliary protein and lipid excretion in rats. Am. J. Physiol. Liver Physiol. 1990, 258, G653–G659. [Google Scholar] [CrossRef]
- Gälman, C.; Angelin, B.; Rudling, M. Bile Acid Synthesis in Humans Has a Rapid Diurnal Variation That Is Asynchronous with Cholesterol Synthesis. Gastroenterol. 2005, 129, 1445–1453. [Google Scholar] [CrossRef]
- Challet, E.; Caldelas, I.; Graff, C.; Pevet, P. Synchronization of the Molecular Clockwork by Light- and Food-Related Cues in Mammals. Boil. Chem. 2003, 384. [Google Scholar] [CrossRef]
- Kamlage, B.; Maldonado, S.G.; Bethan, B.; Peter, E.; Schmitz, O.; Liebenberg, V.; Schatz, P. Quality Markers Addressing Preanalytical Variations of Blood and Plasma Processing Identified by Broad and Targeted Metabolite Profiling. Clin. Chem. 2014, 60, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Van Ravenzwaay, B.; Cunha, G.C.-P.; Leibold, E.; Looser, R.; Mellert, W.; Prokoudine, A.; Walk, T.; Wiemer, J. The use of metabolomics for the discovery of new biomarkers of effect. Toxicol. Lett. 2007, 172, 21–28. [Google Scholar] [CrossRef]
- Van Ravenzwaay, B.; Cunha, G.C.-P.; Strauss, V.; Wiemer, J.; Leibold, E.; Kamp, H.; Walk, T.; Mellert, W.; Looser, R.; Prokoudine, A.; et al. The individual and combined metabolite profiles (metabolomics) of dibutylphthalate and di(2-ethylhexyl)phthalate following a 28-day dietary exposure in rats. Toxicol. Lett. 2010, 198, 159–170. [Google Scholar] [CrossRef]
- Van Ravenzwaay, B.; Herold, M.; Kamp, H.; Kapp, M.; Fabian, E.; Looser, R.; Krennrich, G.; Mellert, W.; Prokoudine, A.; Strauss, V.; et al. Metabolomics: A tool for early detection of toxicological effects and an opportunity for biology based grouping of chemicals—From QSAR to QBAR. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 746, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Roessner, U.; Wagner, C.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 2000, 23, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Walk, T.; Dostler, M. Mass Spectrometry Method for Analyzing Mixtures of Substances. U.S. Patent No. 7,196,323, 27 March 2007. [Google Scholar]
- Walk, T.B.; Looser, R.; Bethan, B.; Herold, M.M.; Kamlage, B.; Schmitz, O.; Wiemer, J.C.; Prokoudine, A.; van Ravenzwaay, B.; Mellert, W.; et al. System and Method for Analyzing a Sample Using Chromatography Coupled Mass Spectrometry. U.S. Patent No. 7,873,481, 18 January 2011. [Google Scholar]
- Behr, C.; Slopianka, M.; Haake, V.; Strauss, V.; Sperber, S.; Kamp, H.; Walk, T.; Beekmann, K.; Rietjens, I.; Van Ravenzwaay, B. Analysis of metabolome changes in the bile acid pool in feces and plasma of antibiotic-treated rats. Toxicol. Appl. Pharmacol. 2019, 363, 79–87. [Google Scholar] [CrossRef]
- Slopianka, M.; Herrmann, A.; Pavkovic, M.; Ellinger-Ziegelbauer, H.; Ernst, R.; Mally, A.; Keck, M.; Riefke, B. Quantitative targeted bile acid profiling as new markers for DILI in a model of methapyrilene-induced liver injury in rats. Toxicology 2017, 386, 1–10. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D. Nlme: Linear and Nonlinear Mixed Effects Models, R package version 3.1-145; R Core Team, 2020; Available online: https://ftp.yz.yamagata-u.ac.jp/pub/cran/web/packages/nlme/ (accessed on 2 June 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Route of Administration | Day | Body Weight | Food Consumption |
|---|---|---|---|---|
| Vancomycin | Gavage | 6 | 0.99 | 0.88 |
| 13 | 0.97 | 0.93 | ||
| 27 | 0.95 | 0.93 | ||
| Intraperitoneal | 6 | 0.98 | 0.91 | |
| 13 | 0.99 | 1.08 | ||
| 27 | 0.99 | 1.20 | ||
| Streptomycin | Gavage | 6 | 0.97 | 0.92 |
| 13 | 0.96 | 0.92 | ||
| 27 | 0.95 | 0.97 | ||
| Subcutaneous | 6 | 1.00 | 0.97 | |
| 13 | 0.99 | 1.02 | ||
| 27 | 1.00 | 1.13 | ||
| Roxithromycin | Gavage | 6 | 0.98 | 0.85 |
| 13 | 0.96 | 0.86 | ||
| 27 | 0.96 | 1.04 | ||
| Intraperitoneal | 6 | 0.97 | 0.92 | |
| 13 | 0.96 | 0.99 | ||
| 27 | 0.97 | 1.05 |
| Treatment | Class of Antibiotics | Route of Administration | Dose/Day (mg/kg Body Weight) | Form of Preparation |
|---|---|---|---|---|
| Vancomycin | Glycopeptide | Gavage | 50 | In ultra-pure water |
| Intraperitoneal | 50 | In saline | ||
| Streptomycin | Aminoglycoside | Gavage | 100 | In drinking water containing 0.5% CMC |
| Subcutaneous | 120 | In saline | ||
| Roxithromycin | Macrolide | Gavage | 200 | In drinking water containing 0.5% CMC |
| Intraperitoneal | 20 | In saline |
| Controls | Vehicle |
|---|---|
| Feeding | None |
| Gavage | 0.5% CMC in drinking water |
| Intraperitoneal | Saline solution |
| Subcutaneous | Saline solution |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bruijn, V.; Behr, C.; Sperber, S.; Walk, T.; Ternes, P.; Slopianka, M.; Haake, V.; Beekmann, K.; van Ravenzwaay, B. Antibiotic-Induced Changes in Microbiome-Related Metabolites and Bile Acids in Rat Plasma. Metabolites 2020, 10, 242. https://doi.org/10.3390/metabo10060242
de Bruijn V, Behr C, Sperber S, Walk T, Ternes P, Slopianka M, Haake V, Beekmann K, van Ravenzwaay B. Antibiotic-Induced Changes in Microbiome-Related Metabolites and Bile Acids in Rat Plasma. Metabolites. 2020; 10(6):242. https://doi.org/10.3390/metabo10060242
Chicago/Turabian Stylede Bruijn, Véronique, Christina Behr, Saskia Sperber, Tilmann Walk, Philipp Ternes, Markus Slopianka, Volker Haake, Karsten Beekmann, and Bennard van Ravenzwaay. 2020. "Antibiotic-Induced Changes in Microbiome-Related Metabolites and Bile Acids in Rat Plasma" Metabolites 10, no. 6: 242. https://doi.org/10.3390/metabo10060242
APA Stylede Bruijn, V., Behr, C., Sperber, S., Walk, T., Ternes, P., Slopianka, M., Haake, V., Beekmann, K., & van Ravenzwaay, B. (2020). Antibiotic-Induced Changes in Microbiome-Related Metabolites and Bile Acids in Rat Plasma. Metabolites, 10(6), 242. https://doi.org/10.3390/metabo10060242