MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics



Abstract
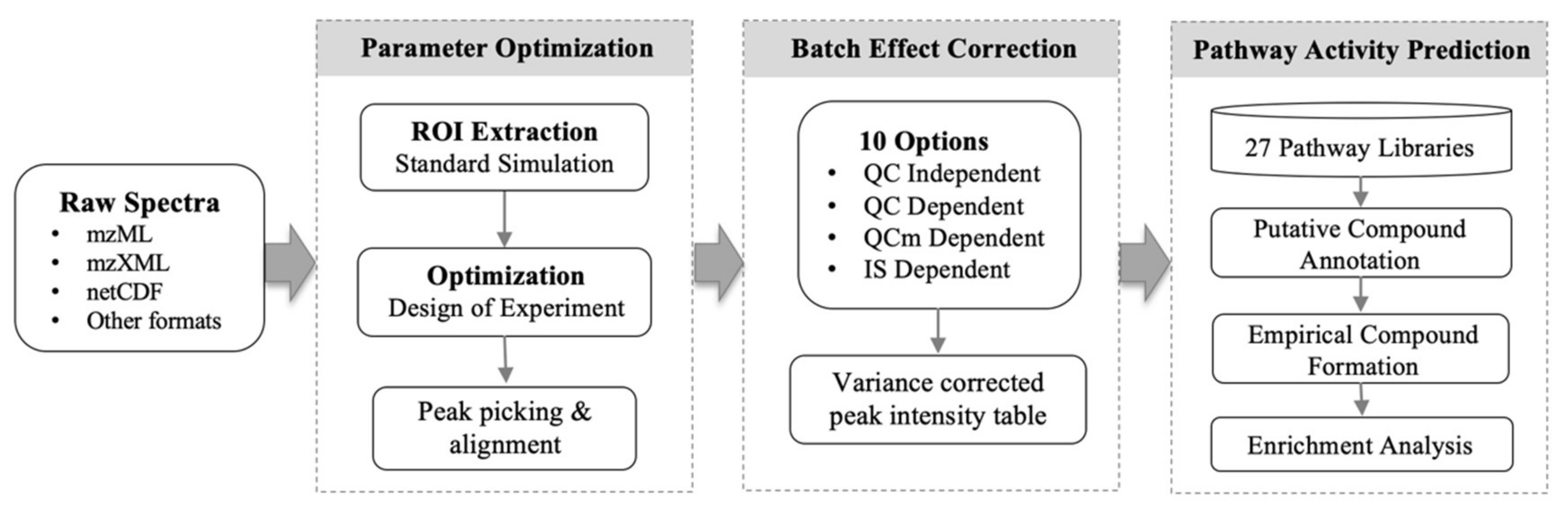
1. Introduction
2. Results
2.1. Peak Identification Benchmark Case Study
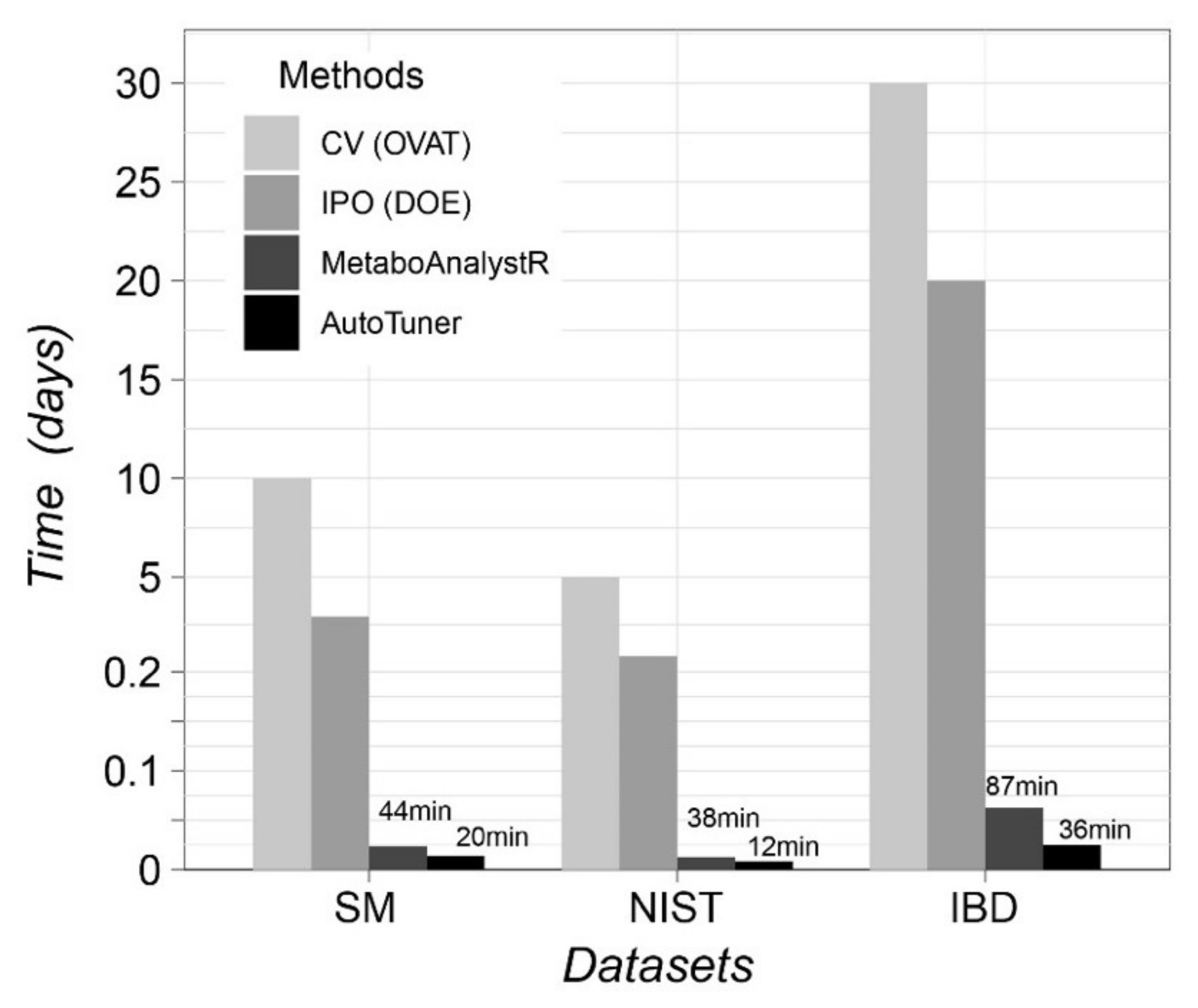
2.2. Algorithm Reliability Benchmark Case Study
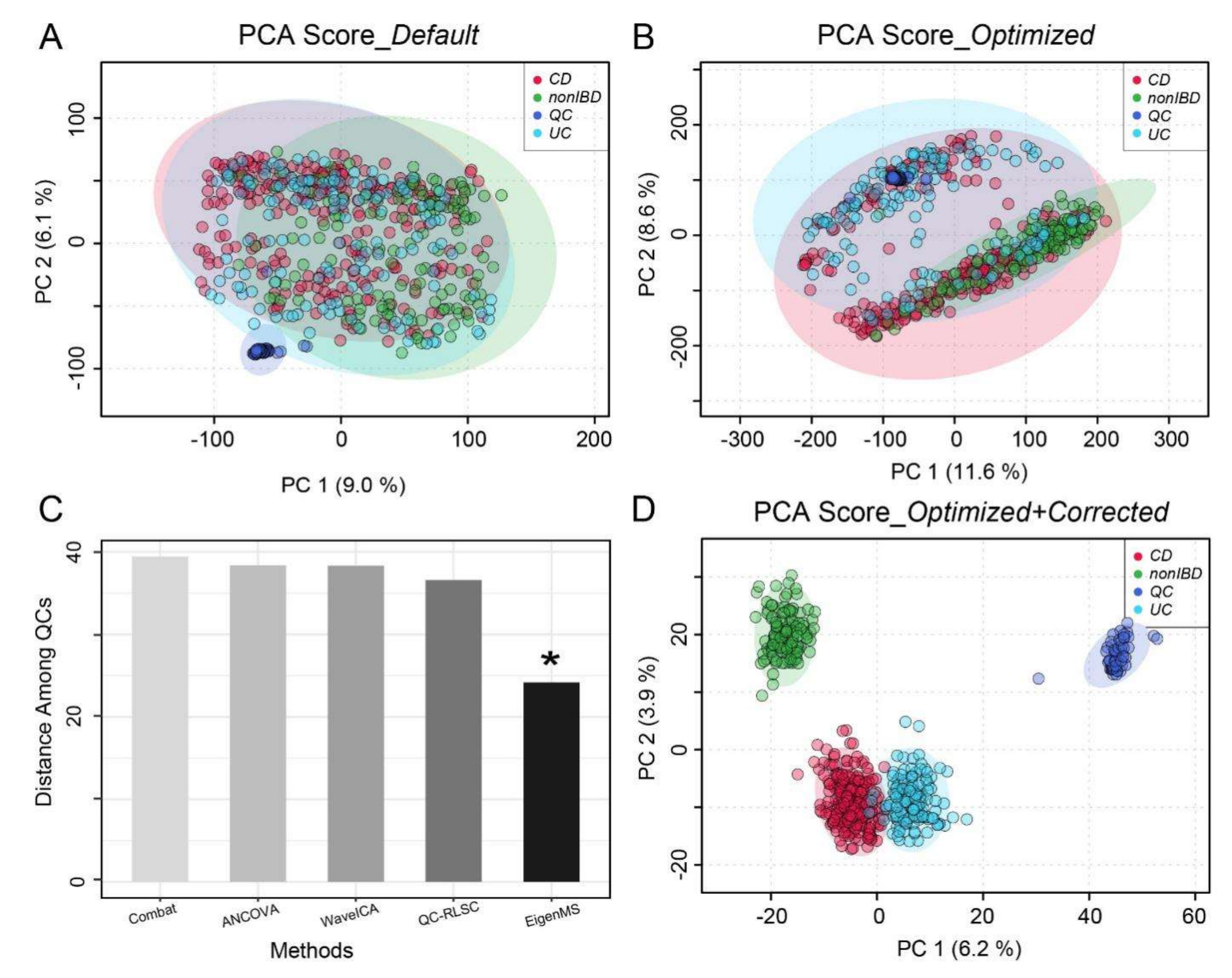
2.3. Overall Workflow Evaluation Using A Large-Scale Clinical Dataset
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Peak Picking Optimization
5.1.1. Extraction of Representative Peaks from Regions of Interest (ROIs)
5.1.2. Design of Experiment (DoE) Based Optimization
5.2. Adaptive Batch Effort Correction
5.3. Mummichog 2 for Pathway Activity Prediction
- (1)
- All m/z features are matched to potential compounds considering isotopes and adducts. Then, per compound, all matching m/z features are split into ECs based on whether they match within an expected retention time window. By default, the retention time window (in seconds) is calculated as the maximum retention time * 0.02. This results in the initial EC list. Users can either customize the retention time fraction (default is 0.02) or retention time tolerance in general in the UpdateInstrumentParameters function (rt_frac and rt_tol, respectively).
- (2)
- ECs are merged if they have the same m/z, matched form/ion, and retention time. This results in the merged empirical compounds list.
- (3)
- Primary ions are enforced (defined in the UpdateInstrumentParameters function [force_primary_ion]), only ECs containing at least one primary ion are kept. Primary ions considered are ‘M+H[1+]’, ‘M+Na[1+]’, ‘M−H2O+H[1+]’, ‘M−H[−]’, ‘M−2H[2−]’, ‘M−H2O−H[−]’, ‘M+H [1+]’, ‘M+Na [1+]’, ‘M−H2O+H [1+]’, ‘M−H [1−]’, ‘M−2H [2−]’, and ‘M−H2O−H[1−]’. This produces the final EC list.
- (4)
- Pathway libraries are converted from “Compound” space to “Empirical Compound” space. This is done by converting all compounds in each pathway to all empirical compound matches. Then, the mummichog/GSEA algorithm works as before to calculate pathway enrichment.
- (5)
- To use the updated algorithm, set the version parameter in SetPeakEnrichMethod to “v2”.
5.4. Benchmark Case Studies
5.4.1. Known Standards Mixture
5.4.2. NIST-1950 Serum Diluted Series
5.4.3. Clinical Inflammatory Bowel Disease Data
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartl, J.; Kiefer, P.; Kaczmarczyk, A.; Mittelviefhaus, M.; Meyer, F.; Vonderach, T.; Hattendorf, B.; Jenal, U.; Vorholt, J.A. Untargeted metabolomics links glutathione to bacterial cell cycle progression. Nat. Metab. 2020, 2, 153–166. [Google Scholar] [CrossRef]
- Garza, D.R.; Van Verk, M.C.; Huynen, M.A.; Dutilh, B.E. Towards predicting the environmental metabolome from metagenomics with a mechanistic model. Nat. Microbiol. 2018, 3, 456–460. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, N.D.; Watrous, J.; A Kapono, C.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Uppal, K.; Walker, D.I.; Liu, K.; Li, S.; Go, Y.-M.; Jones, D.P. Computational Metabolomics: A Framework for the Million Metabolome. Chem. Res. Toxicol. 2016, 29, 1956–1975. [Google Scholar] [CrossRef]
- Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolities 2019, 9, 57. [Google Scholar] [CrossRef]
- De Bruycker, K.; Welle, A.; Hirth, S.; Blanksby, S.J.; Barner-Kowollik, C. Mass spectrometry as a tool to advance polymer science. Nat. Rev. Chem. 2020, 1–12. [Google Scholar] [CrossRef]
- Albóniga, O.E.; González, O.; Alonso, R.M.; Xu, Y.; Goodacre, R. Optimization of XCMS parameters for LC–MS metabolomics: An assessment of automated versus manual tuning and its effect on the final results. Metabolomics 2020, 16, 14. [Google Scholar] [CrossRef]
- Nash, W.; Dunn, W.B. From mass to metabolite in human untargeted metabolomics: Recent advances in annotation of metabolites applying liquid chromatography-mass spectrometry data. TrAC Trends Anal. Chem. 2019, 120, 115324. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Libiseller, G.; Dvorzak, M.; Kleb, U.; Gander, E.; Eisenberg, T.; Madeo, F.; Neumann, S.; Trausinger, G.; Sinner, F.; Pieber, T.; et al. IPO: A tool for automated optimization of XCMS parameters. BMC Bioinform. 2015, 16, 736. [Google Scholar] [CrossRef]
- McLean, C.; Kujawinski, E.B. AutoTuner: High Fidelity and Robust Parameter Selection for Metabolomics Data Processing. Anal. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Clausen, M.R.; Dalsgaard, T.K.; Mortensen, G.; Bertram, H. Time-Saving Design of Experiment Protocol for Optimization of LC-MS Data Processing in Metabolomic Approaches. Anal. Chem. 2013, 85, 7109–7116. [Google Scholar] [CrossRef]
- Manier, S.K.; Keller, A.; Meyer, M.R. Automated optimization of XCMS parameters for improved peak picking of liquid chromatography-mass spectrometry data using the coefficient of variation and parameter sweeping for untargeted metabolomics. Drug Test. Anal. 2018, 11, 752–761. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; IBDMDB Investigators; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelená, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.; Halsall, A.; The Human Serum Metabolome (HUSERMET) Consortium; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Li, B.; Tang, J.; Yang, Q.; Li, S.; Cui, X.; Li, Y.; Chen, Y.; Xue, W.W.; Li, X.; Zhu, F. NOREVA: Normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017, 45, W162–W170. [Google Scholar] [CrossRef]
- Deng, K.; Zhang, F.; Tan, Q.; Huang, Y.; Song, W.; Rong, Z.; Zhu, Z.-J.; Li, K.; Li, Z. WaveICA: A novel algorithm to remove batch effects for large-scale untargeted metabolomics data based on wavelet analysis. Anal. Chim. Acta 2019, 1061, 60–69. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Benton, H.P.; Siuzdak, G. Annotation: A Computational Solution for Streamlining Metabolomics Analysis. Anal. Chem. 2017, 90, 480–489. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; E Wheelock, C. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.H.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting Network Activity from High Throughput Metabolomics. PLoS Comput. Boil. 2013, 9, e1003123. [Google Scholar] [CrossRef]
- Shuzhao, L. Mummichog. Available online: https://github.com/shuzhao-li/mummichog (accessed on 1 March 2020).
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinform. 2018, 34, 4313–4314. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metab. 2020, 10, 186. [Google Scholar] [CrossRef]
- Li, Z.; Lu, Y.; Guo, Y.; Cao, H.; Wang, Q.; Shui, W. Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Anal. Chim. Acta 2018, 1029, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Simón-Manso, Y.; Lowenthal, M.S.; Kilpatrick, L.E.; Sampson, M.; Telu, K.H.; Rudnick, P.A.; Mallard, W.G.; Bearden, D.W.; Schock, T.B.; Tchekhovskoi, D.V.; et al. Metabolite Profiling of a NIST Standard Reference Material for Human Plasma (SRM 1950): GC-MS, LC-MS, NMR, and Clinical Laboratory Analyses, Libraries, and Web-Based Resources. Anal. Chem. 2013, 85, 11725–11731. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, M.; Rännar, S.; Madsen, R.B.; Donten, M.A.; Marsden-Edwards, E.; Moritz, T.; Shockcor, J.P.; Johansson, E.; Trygg, J. Strategy for Optimizing LC-MS Data Processing in Metabolomics: A Design of Experiments Approach. Anal. Chem. 2012, 84, 6869–6876. [Google Scholar] [CrossRef] [PubMed]
- Cantero, J.M.B.; Flores, E.I.; Alcalde, B.G.; Ortega, E.M.; Muret, F.R.M.; Asenjo, E.C.; Casas, J.A.V. Bile acid malabsorption in patients with chronic diarrhea and Crohn’s disease. Revista Española de Enfermedades Digestivas 2018, 111, 40–45. [Google Scholar] [CrossRef]
- Uchiyama, K.; Kishi, H.; Komatsu, W.; Nagao, M.; Ohhira, S.; Kobashi, G. Lipid and Bile Acid Dysmetabolism in Crohn’s Disease. J. Immunol. Res. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Kuroki, F.; Iida, M.; Tominaga, M.; Matsumoto, T.; Kanamoto, K.; Fujishima, M. Is vitamin E depleted in Crohn’s disease at initial diagnosis? Dig. Dis. 1994, 12, 248–254. [Google Scholar] [CrossRef]
- Narula, N.; Cooray, M.; Anglin, R.; Muqtadir, Z.; Narula, A.; Marshall, J.K. Impact of High-Dose Vitamin D3 Supplementation in Patients with Crohn’s Disease in Remission: A Pilot Randomized Double-Blind Controlled Study. Dig. Dis. Sci. 2016, 62, 448–455. [Google Scholar] [CrossRef]
- Dionne, S.; Calderon, M.R.; White, J.H.; Memari, B.; Elimrani, I.; Adelson, B.; Piccirillo, C.; Seidman, E.G. Differential effect of vitamin D on NOD2- and TLR-induced cytokines in Crohn’s disease. Mucosal Immunol. 2014, 7, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Scoville, E.A.; Allaman, M.M.; Brown, C.T.; Motley, A.K.; Horst, S.N.; Williams, C.S.; Koyama, T.; Zhao, Z.; Adams, D.W.; Beaulieu, D.B.; et al. Alterations in lipid, amino acid, and energy metabolism distinguish Crohn’s disease from ulcerative colitis and control subjects by serum metabolomic profiling. Metabolomics 2017, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Kolacek, M.; Paduchova, Z.; Dvorakova, M.; Zitnanova, I.; Cierna, I.; Durackova, Z.; Muchova, J. Effect of natural polyphenols on thromboxane levels in children with Crohn’s disease. Bratisl. Med J. 2019, 120, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Petrey, A.C.; De La Motte, C.A. Hyaluronan in inflammatory bowel disease: Cross-linking inflammation and coagulation. Matrix Boil. 2019, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Nikolic, S.B.; Wilson, R.; Sharman, J.E.; Edwards, L.M. Metabolomics Data Normalization with EigenMS. PLoS ONE 2014, 9, e116221. [Google Scholar] [CrossRef]
- Wehrens, R.; Hageman, J.A.; Van Eeuwijk, F.; Kooke, R.; Flood, P.J.; Wijnker, E.; Keurentjes, J.J.; Lommen, A.; Van Eekelen, H.D.L.M.; Hall, R.D.; et al. Improved batch correction in untargeted MS-based metabolomics. Metabolomics 2016, 12, 88. [Google Scholar] [CrossRef]
- De Livera, A.M.; Sysi-Aho, M.; Jacob, L.; Gagnon-Bartsch, J.A.; Castillo, S.; Simpson, J.A.; Speed, T.P. Statistical Methods for Handling Unwanted Variation in Metabolomics Data. Anal. Chem. 2015, 87, 3606–3615. [Google Scholar] [CrossRef]
- De Livera, A.M.; Dias, D.A.; De Souza, D.P.; Rupasinghe, T.; Pyke, J.; Tull, D.; Roessner, U.; McConville, M.; Speed, T.P. Normalizing and Integrating Metabolomics Data. Anal. Chem. 2012, 84, 10768–10776. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Sysi-Aho, M.; Katajamaa, M.; Yetukuri, L.; Orešič, M. Normalization method for metabolomics data using optimal selection of multiple internal standards. BMC Bioinform. 2007, 8, 93. [Google Scholar] [CrossRef]
- Redestig, H.; Fukushima, A.; Stenlund, H.; Moritz, T.; Arita, M.; Saito, K.; Kusano, M. Compensation for Systematic Cross-Contribution Improves Normalization of Mass Spectrometry Based Metabolomics Data. Anal. Chem. 2009, 81, 7974–7980. [Google Scholar] [CrossRef] [PubMed]
- Mahieu, N.G.; Patti, G.J. Systems-Level Annotation of a Metabolomics Data Set Reduces 25 000 Features to Fewer than 1000 Unique Metabolites. Anal. Chem. 2017, 89, 10397–10406. [Google Scholar] [CrossRef]
- Chambers, M.C.; MacLean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Total Peaks | True Peaks | Quantified Consensus | Gaussian Peak Ratio |
|---|---|---|---|---|
| Default | 16,896 | 382 | 350 | 47.8% |
| IPO | 24,346 | 744 | 663 | 52.0% |
| AutoTuner | 25,517 | 664 | 603 | 40.5% |
| MetaboAnalystR 3.0 | 18,044 | 799 | 754 | 64.4% |
| Mummichog v1.0.8 | Mummichog v2.0 | ||
|---|---|---|---|
| Pathways | p Value | Pathways | p Value |
| Bile acid biosynthesis | 0.017199 | Bile acid biosynthesis | 0.011283 |
| Vitamin D3 (cholecalciferol) metabolism | 0.017526 | Vitamin E metabolism | 0.011321 |
| Vitamin E metabolism | 0.017966 | Vitamin D3 (cholecalciferol) metabolism | 0.014207 |
| Carnitine shuttle | 0.018084 | Galactose metabolism | 0.016026 |
| Glycosphingolipid metabolism | 0.021048 | Glycerophospholipid metabolism | 0.020464 |
| De novo fatty acid biosynthesis | 0.026554 | Carnitine shuttle | 0.021085 |
| Keratan sulfate degradation | 0.031317 | Chondroitin sulfate degradation | 0.025739 |
| Fatty Acid Metabolism | 0.032132 | Vitamin B2 (riboflavin) metabolism | 0.025739 |
| N-Glycan Degradation | 0.043912 | Vitamin H (biotin) metabolism | 0.025739 |
| Phosphatidylinositol phosphate metabolism | 0.053756 | Fatty acid oxidation | 0.025739 |
| Hexose phosphorylation | 0.069236 | Omega-6 fatty acid metabolism | 0.025739 |
| Fatty acid activation | 0.075044 | Glycosphingolipid metabolism | 0.041115 |
| Limonene and pinene degradation | 0.078492 | Phosphatidylinositol phosphate metabolism | 0.043604 |
| Chondroitin sulfate degradation | 0.082534 | Hyaluronan Metabolism | 0.04815 |
| Glycosphingolipid biosynthesis - globoseries | 0.082534 | Putative anti-Inflammatory metabolites formation from EPA | 0.04815 |
| Saturated fatty acids beta-oxidation | 0.082534 | Electron transport chain | 0.04815 |
| Heparan sulfate degradation | 0.082534 | Heparan sulfate degradation | 0.04815 |
| Glycerophospholipid metabolism | 0.09418 | Sialic acid metabolism | 0.061564 |
| Starch and Sucrose Metabolism | 0.13566 | Vitamin A (retinol) metabolism | 0.061564 |
| Ascorbate (Vitamin C) and Aldarate Metabolism | 0.14503 | Saturated fatty acids beta-oxidation | 0.061564 |
| Categories | Methods |
|---|---|
| QC Sample Independent | Combat [37], WaveICA [18], Eigens MS [38] |
| QC Sample Dependent | QC-RLSC [16], ANCOVA [39] |
| QC Metabolite Dependent | RUV-random [40], RUV2 [41], RUVseq [42] |
| Internal Standards Dependent | NOMIS [43], CCMN [44] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. https://doi.org/10.3390/metabo10050186
Pang Z, Chong J, Li S, Xia J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites. 2020; 10(5):186. https://doi.org/10.3390/metabo10050186
Chicago/Turabian StylePang, Zhiqiang, Jasmine Chong, Shuzhao Li, and Jianguo Xia. 2020. "MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics" Metabolites 10, no. 5: 186. https://doi.org/10.3390/metabo10050186
APA StylePang, Z., Chong, J., Li, S., & Xia, J. (2020). MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites, 10(5), 186. https://doi.org/10.3390/metabo10050186