Bile Acid Profile and its Changes in Response to Cefoperazone Treatment in MR1 Deficient Mice

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
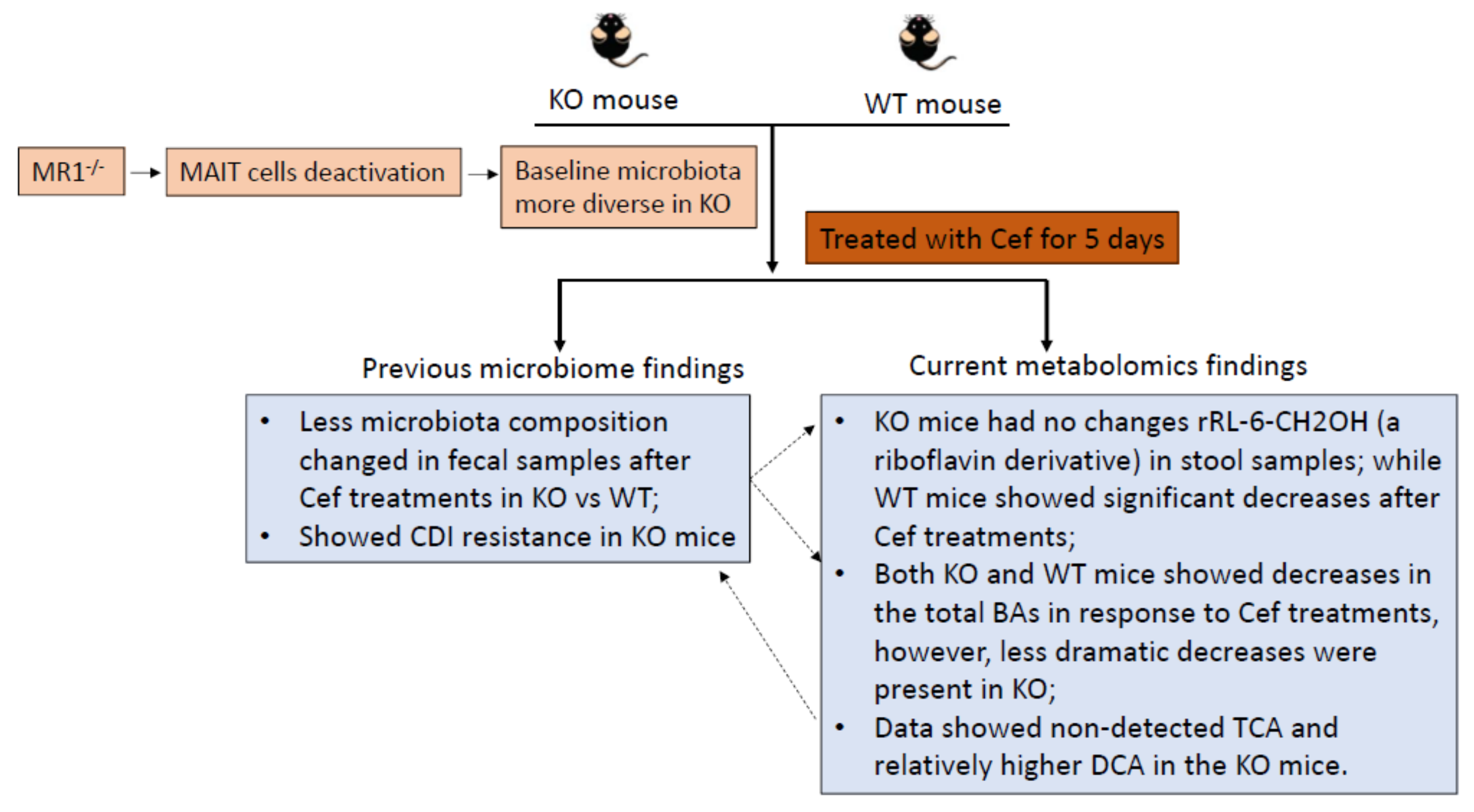{kind=link}
Abstract
1. Introduction
2. Results
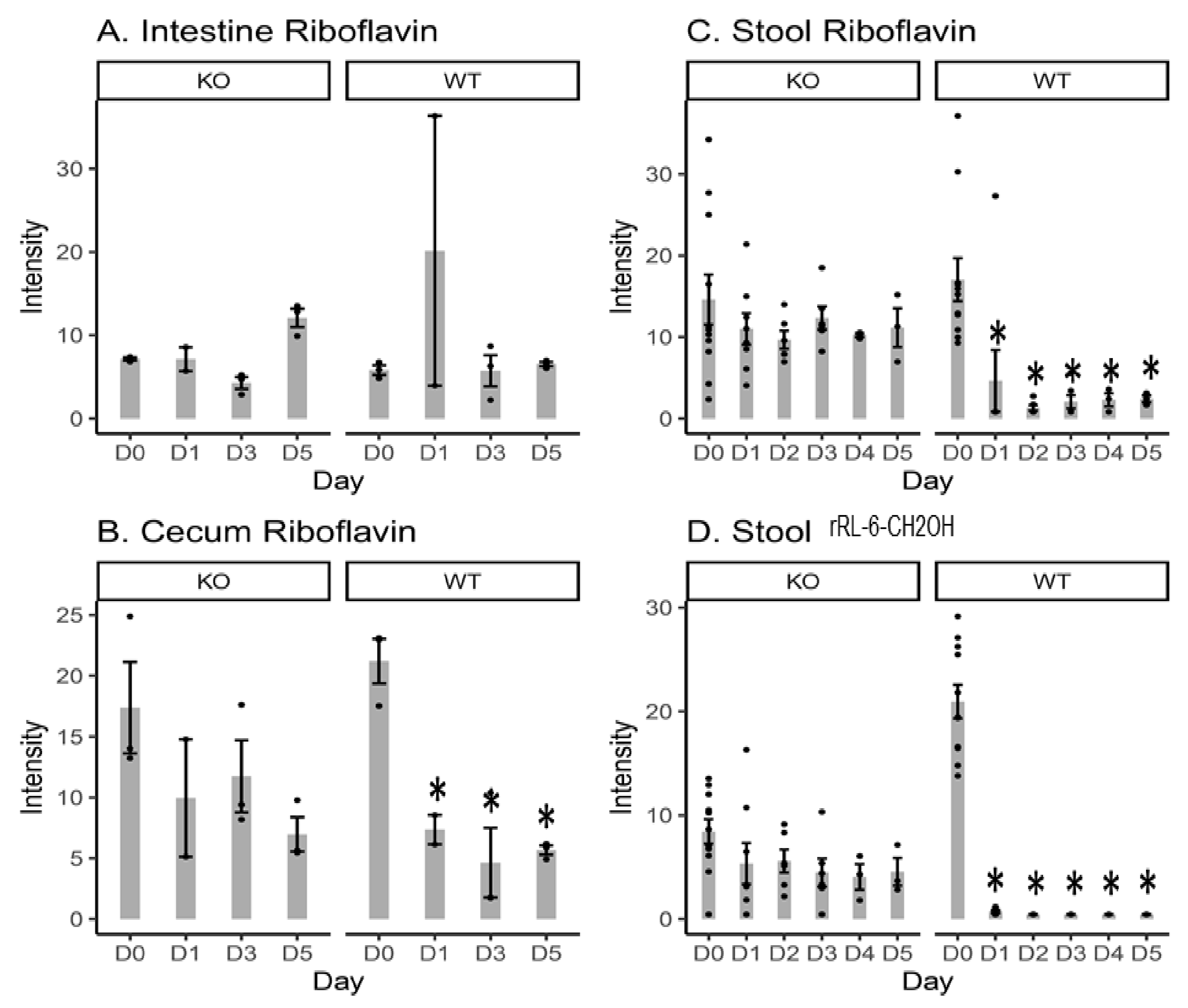
2.1. Riboflavin and Bacteria-Biosynthesized Riboflavin Metabolite Intensity Levels
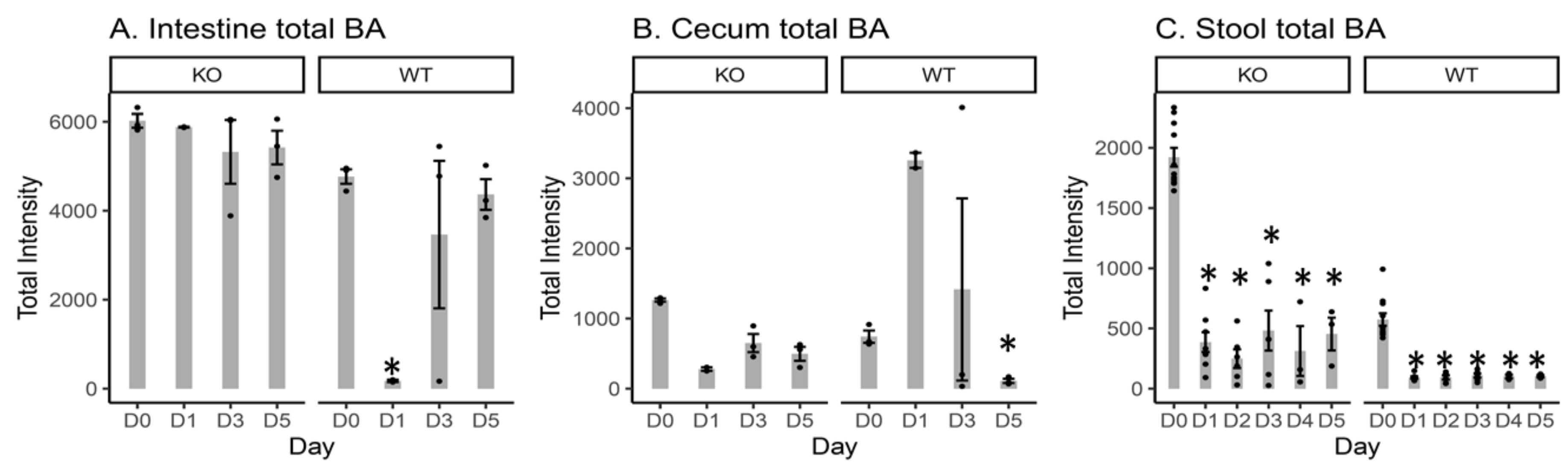
2.2. Total Measured BA Intensity Levels in the Intestinal Content, Cecal Content, and Stool Samples from MR1−/− KO and WT Mice
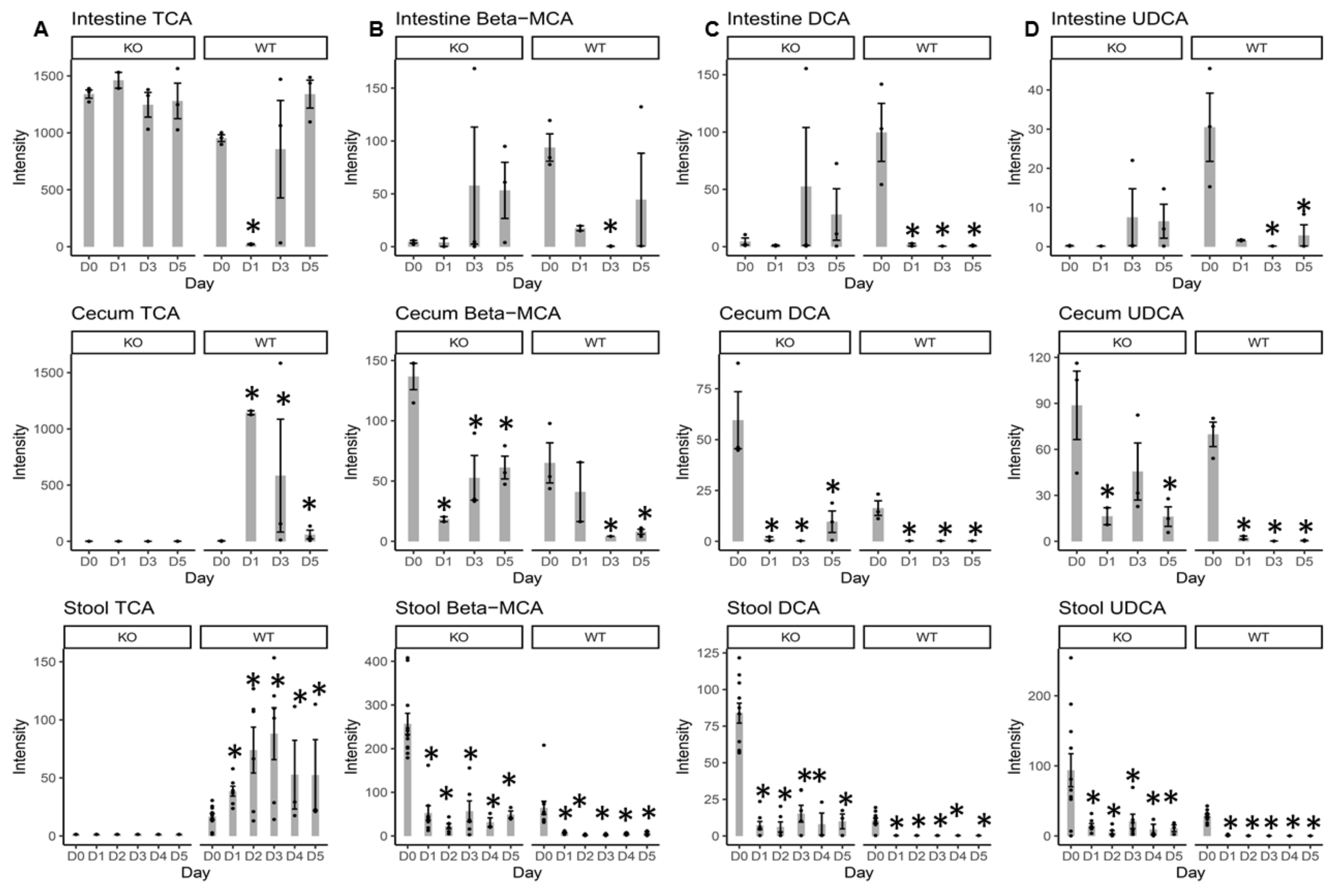
2.3. Individual BA Intensity Levels in the Intestinal Content, Cecal Content, and Stool Samples from MR1−/− KO and WT Mice
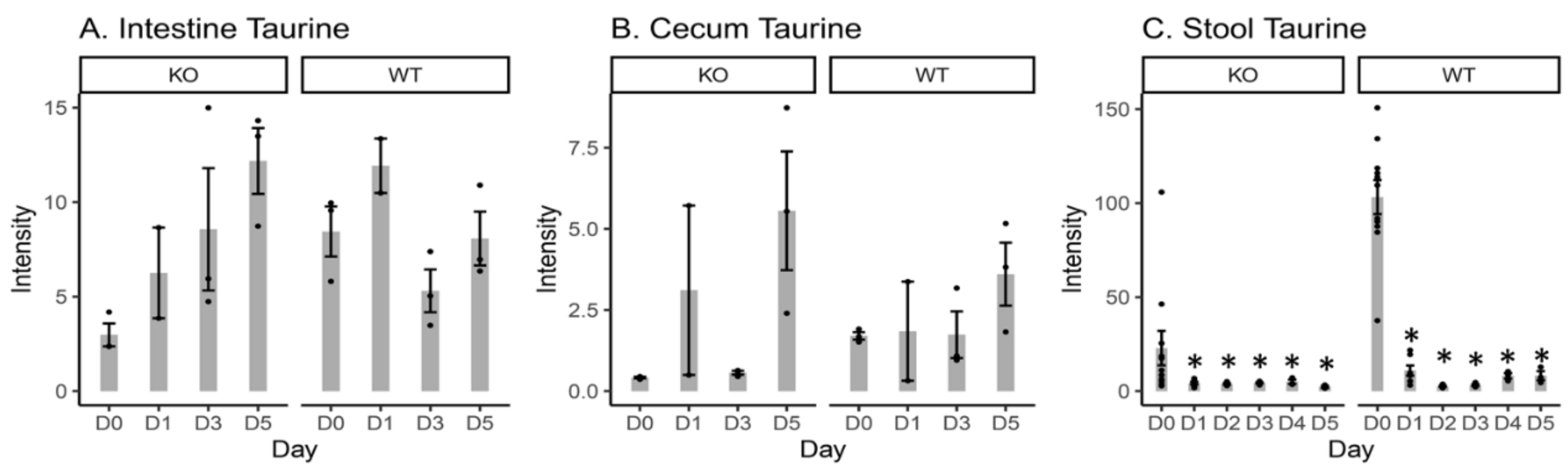
2.4. Free Taurine Levels in the Intestinal Content, Cecal Content, and Stool Samples
3. Discussion
4. Material and Methods
4.1. Chemicals
4.2. Animal Care and Treatment
4.3. Quality Control in Metabolomics
4.4. Open Metabolic Profiling by UPLC/QTof-MS
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Disclaimer
Abbreviations
References
- Gherardin, N.A.; Keller, A.N.; Woolley, R.E.; Le Nours, J.; Ritchie, D.S.; Neeson, P.J.; Birkinshaw, R.W.; Eckle, S.B.G.; Waddington, J.N.; Liu, L.; et al. Diversity of T Cells Restricted by the MHC Class I-Related Molecule MR1 Facilitates Differential Antigen Recognition. Immunity 2016, 44, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Martin, E.; Kim, S.; Yu, L.; Soudais, C.; Fremont, D.H.; Lantz, O.; Hansen, T.H. MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8290–8295. [Google Scholar] [CrossRef] [PubMed]
- Le Bourhis, L.; Martin, E.; Peguillet, I.; Guihot, A.; Froux, N.; Core, M.; Levy, E.; Dusseaux, M.; Meyssonnier, V.; Premel, V.; et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol. 2010, 11, 701–708. [Google Scholar] [CrossRef]
- Davanian, H.; Gaiser, R.A.; Silfverberg, M.; Hugerth, L.W.; Sobkowiak, M.J.; Lu, L.; Healy, K.; Sandberg, J.K.; Nasman, P.; Karlsson, J.; et al. Mucosal-associated invariant T cells and oral microbiome in persistent apical periodontitis. Int. J. Oral. Sci. 2019, 11, 16. [Google Scholar] [CrossRef]
- Booth, J.S.; Salerno-Goncalves, R.; Blanchard, T.G.; Patil, S.A.; Kader, H.A.; Safta, A.M.; Morningstar, L.M.; Czinn, S.J.; Greenwald, B.D.; Sztein, M.B. Mucosal-Associated Invariant T Cells in the Human Gastric Mucosa and Blood: Role in Helicobacter pylori Infection. Front. Immunol. 2015, 6, 466. [Google Scholar] [CrossRef]
- Chiba, A.; Tajima, R.; Tomi, C.; Miyazaki, Y.; Yamamura, T.; Miyake, S. Mucosal-associated invariant T cells promote inflammation and exacerbate disease in murine models of arthritis. Arthritis Rheum. 2012, 64, 153–161. [Google Scholar] [CrossRef]
- Kwon, Y.S.; Cho, Y.N.; Kim, M.J.; Jin, H.M.; Jung, H.J.; Kang, J.H.; Park, K.J.; Kim, T.J.; Kee, H.J.; Kim, N.; et al. Mucosal-associated invariant T cells are numerically and functionally deficient in patients with mycobacterial infection and reflect disease activity. Tuberculosis 2015, 95, 267–274. [Google Scholar] [CrossRef]
- Meierovics, A.I.; Cowley, S.C. MAIT cells promote inflammatory monocyte differentiation into dendritic cells during pulmonary intracellular infection. J. Exp. Med. 2016, 213, 2793–2809. [Google Scholar] [CrossRef]
- Loh, L.; Wang, Z.; Sant, S.; Koutsakos, M.; Jegaskanda, S.; Corbett, A.J.; Liu, L.; Fairlie, D.P.; Crowe, J.; Rossjohn, J.; et al. Human mucosal-associated invariant T cells contribute to antiviral influenza immunity via IL-18-dependent activation. Proc. Natl. Acad. Sci. USA 2016, 113, 10133–10138. [Google Scholar] [CrossRef]
- Smith, A.D.; Zhang, I.T.; Schubert, A.M.; Giordano, N.P.; Hastie, J.E.; Cowley, S.C.; Carlson, P.E. MAIT cells: Shaping the microbiome, contributing to Clostridium difficile infection. J. Immunol. 2017, 198, 216. [Google Scholar]
- Smith, A.D.; Foss, E.D.; Zhang, I.; Hastie, J.L.; Giordano, N.P.; Gasparyan, L.; VinhNguyen, L.P.; Schubert, A.M.; Prasad, D.; McMichael, H.L.; et al. Microbiota of MR1 deficient mice confer resistance against Clostridium difficile infection. PLoS ONE 2019, 14, e0223025. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Dore, J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel. Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Thompson-Chagoyan, O.C.; Maldonado, J.; Gil, A. Colonization and impact of disease and other factors on intestinal microbiota. Dig. Dis. Sci. 2007, 52, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Sun, J.; Schnackenberg, L.K.; Khare, S.; Yang, X.; Greenhaw, J.; Salminen, W.; Mendrick, D.L.; Beger, R.D. Evaluating effects of penicillin treatment on the metabolome of rats. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 932, 134–143. [Google Scholar] [CrossRef]
- Antunes, L.C.; Han, J.; Ferreira, R.B.; Lolic, P.; Borchers, C.H.; Finlay, B.B. Effect of antibiotic treatment on the intestinal metabolome. Antimicrob. Agents Chemother. 2011, 55, 1494–1503. [Google Scholar] [CrossRef]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Jun, Z.L.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef]
- Yap, I.K.; Li, J.V.; Saric, J.; Martin, F.P.; Davies, H.; Wang, Y.; Wilson, I.D.; Nicholson, J.K.; Utzinger, J.; Marchesi, J.R.; et al. Metabonomic and microbiological analysis of the dynamic effect of vancomycin-induced gut microbiota modification in the mouse. J. Proteome. Res. 2008, 7, 3718–3728. [Google Scholar] [CrossRef]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. mSphere 2016, 1, e00045–15. [Google Scholar] [CrossRef]
- Kochan, T.J.; Somers, M.J.; Kaiser, A.M.; Shoshiev, M.S.; Hagan, A.K.; Hastie, J.L.; Giordano, N.P.; Smith, A.D.; Schubert, A.M.; Carlson, P.E., Jr.; et al. Intestinal calcium and bile salts facilitate germination of Clostridium difficile spores. PLoS Pathog. 2017, 13, e1006443. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.K.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Jamma, T.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, D.A.; Huse, S.M.; Morrison, H.G.; Schmidt, T.M.; Sogin, M.L.; Young, V.B. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect. Immun. 2009, 77, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Hosomi, K.; Sawane, K.; Kunisawa, J. Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front. Nutr. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Gika, H.G.; Zisi, C.; Theodoridis, G.; Wilson, I.D. Protocol for quality control in metabolic profiling of biological fluids by U(H)PLC-MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1008, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Von Tungeln, L.S.; Hines, W.; Beger, R.D. Identification of metabolite profiles of the catechol-O-methyl transferase inhibitor tolcapone in rat urine using LC/MS-based metabonomics analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2557–2565. [Google Scholar] [CrossRef]
- Sun, J.; Schnackenberg, L.K.; Hansen, D.K.; Beger, R.D. Study of valproic acid-induced endogenous and exogenous metabolite alterations using LC-MS-based metabolomics. Bioanalysis 2010, 2, 207–216. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucl. Acids Res 2007, 35, D521–526. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. 3.3.1 ed.; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Cao, Z.; Smith, A.D.; Carlson Jr, P.E.; Coryell, M.; Chen, H.; Beger, R.D. Bile Acid Profile and its Changes in Response to Cefoperazone Treatment in MR1 Deficient Mice. Metabolites 2020, 10, 127. https://doi.org/10.3390/metabo10040127
Sun J, Cao Z, Smith AD, Carlson Jr PE, Coryell M, Chen H, Beger RD. Bile Acid Profile and its Changes in Response to Cefoperazone Treatment in MR1 Deficient Mice. Metabolites. 2020; 10(4):127. https://doi.org/10.3390/metabo10040127
Chicago/Turabian StyleSun, Jinchun, Zhijun Cao, Ashley D. Smith, Paul E. Carlson Jr, Michael Coryell, Huizhong Chen, and Richard D. Beger. 2020. "Bile Acid Profile and its Changes in Response to Cefoperazone Treatment in MR1 Deficient Mice" Metabolites 10, no. 4: 127. https://doi.org/10.3390/metabo10040127
APA StyleSun, J., Cao, Z., Smith, A. D., Carlson Jr, P. E., Coryell, M., Chen, H., & Beger, R. D. (2020). Bile Acid Profile and its Changes in Response to Cefoperazone Treatment in MR1 Deficient Mice. Metabolites, 10(4), 127. https://doi.org/10.3390/metabo10040127