Intracellular Metabolites in Marine Microorganisms during an Experiment Evaluating Microbial Mortality

Abstract
1. Introduction
2. Results
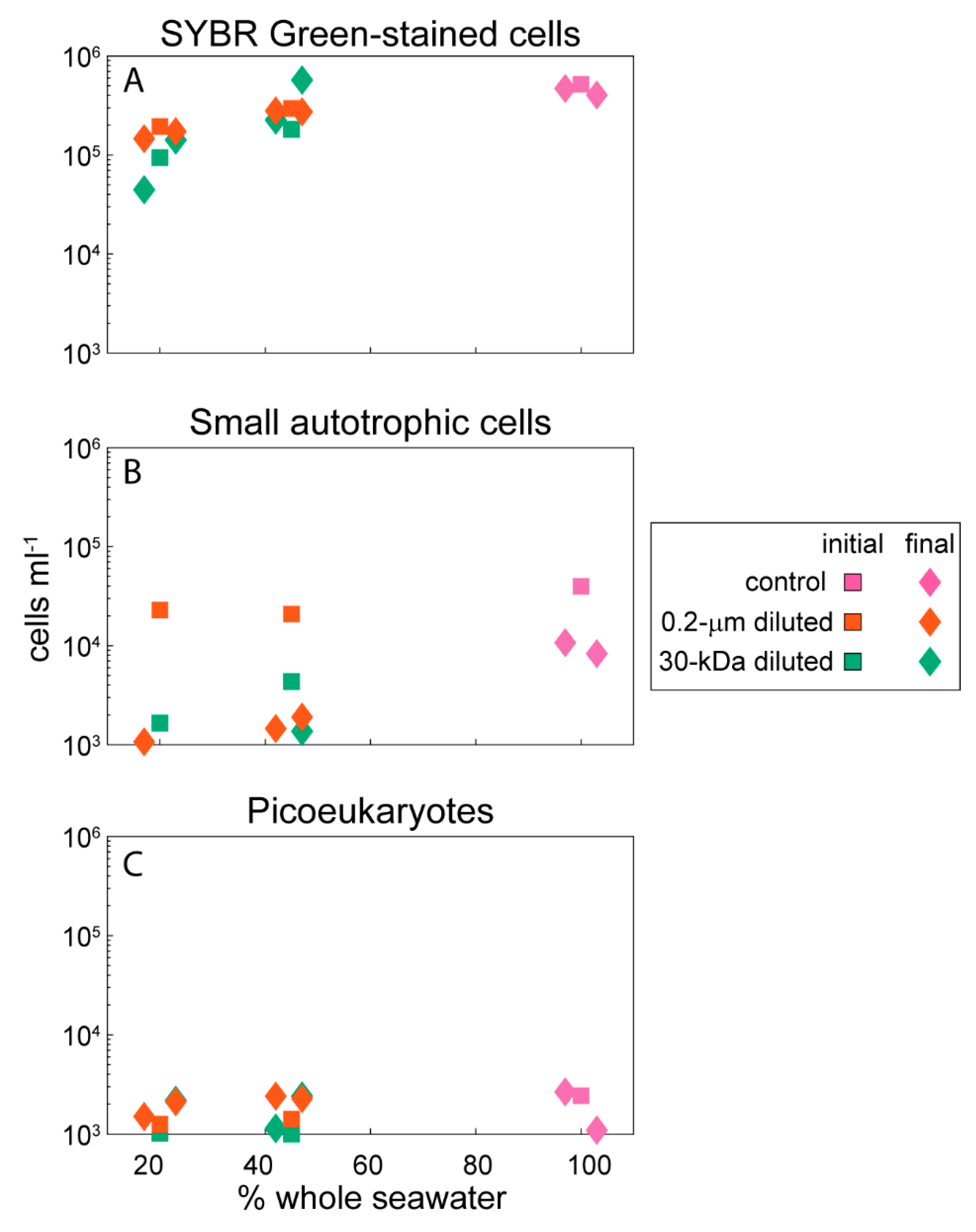
2.1. Environmental Data
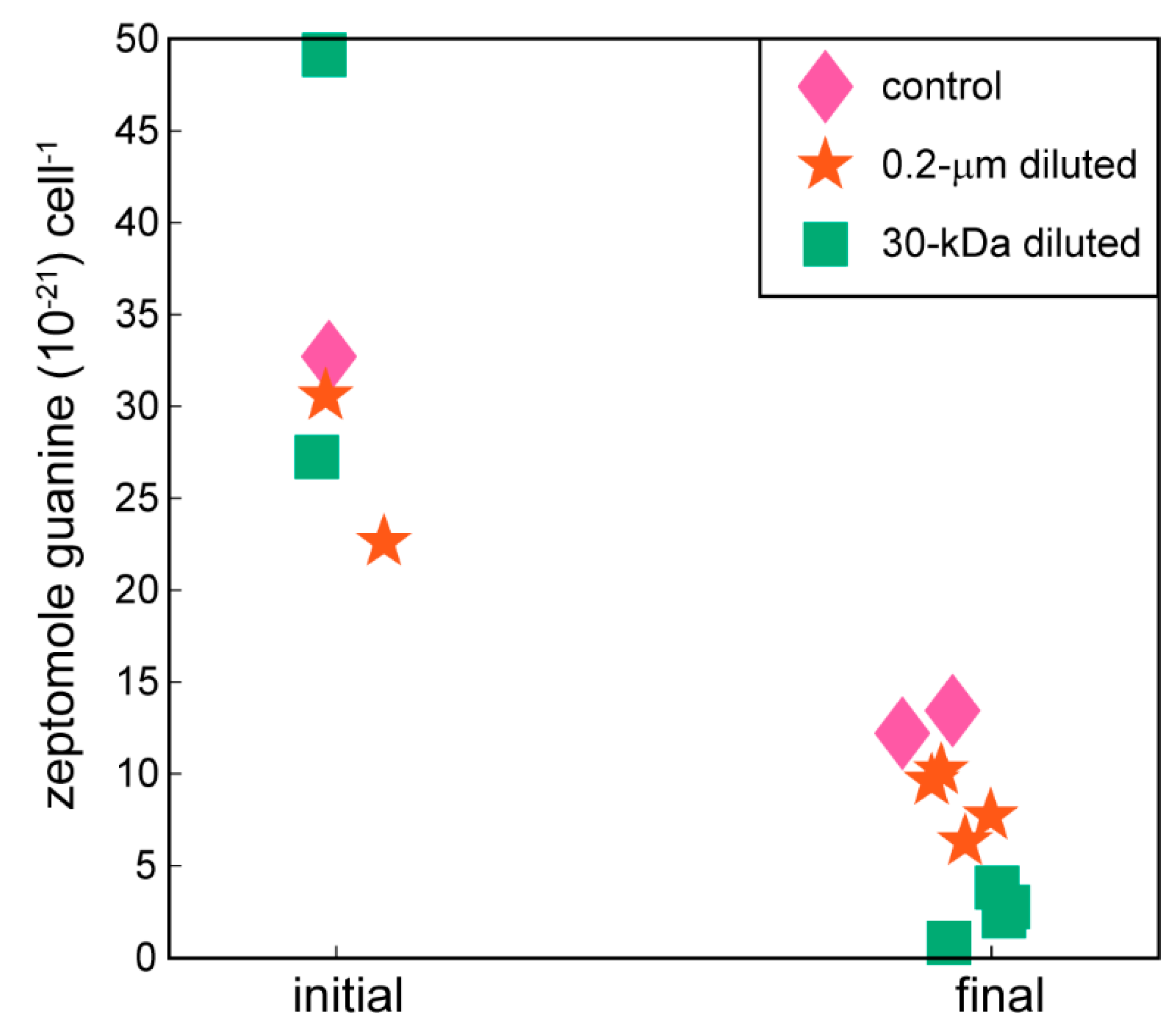
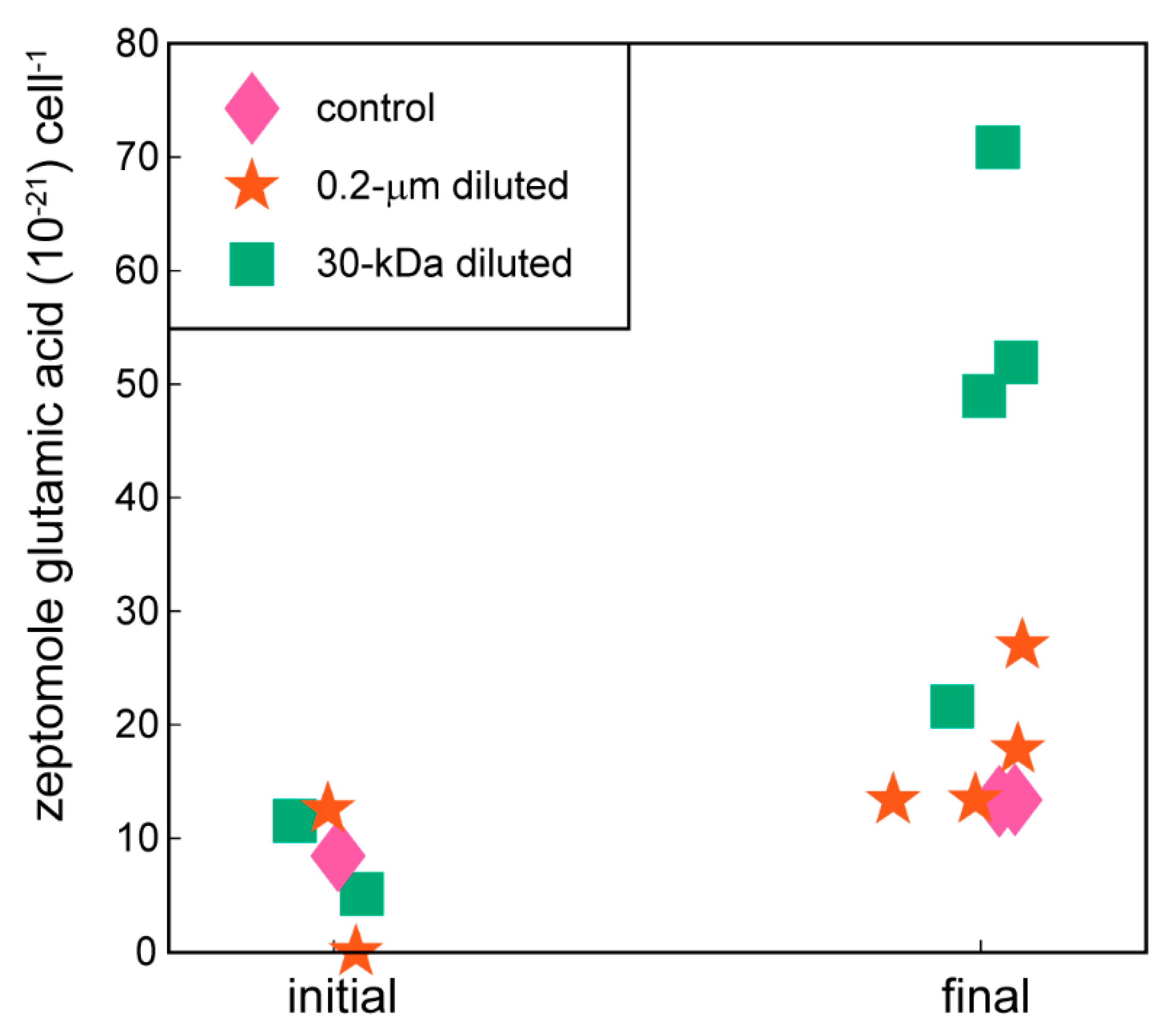
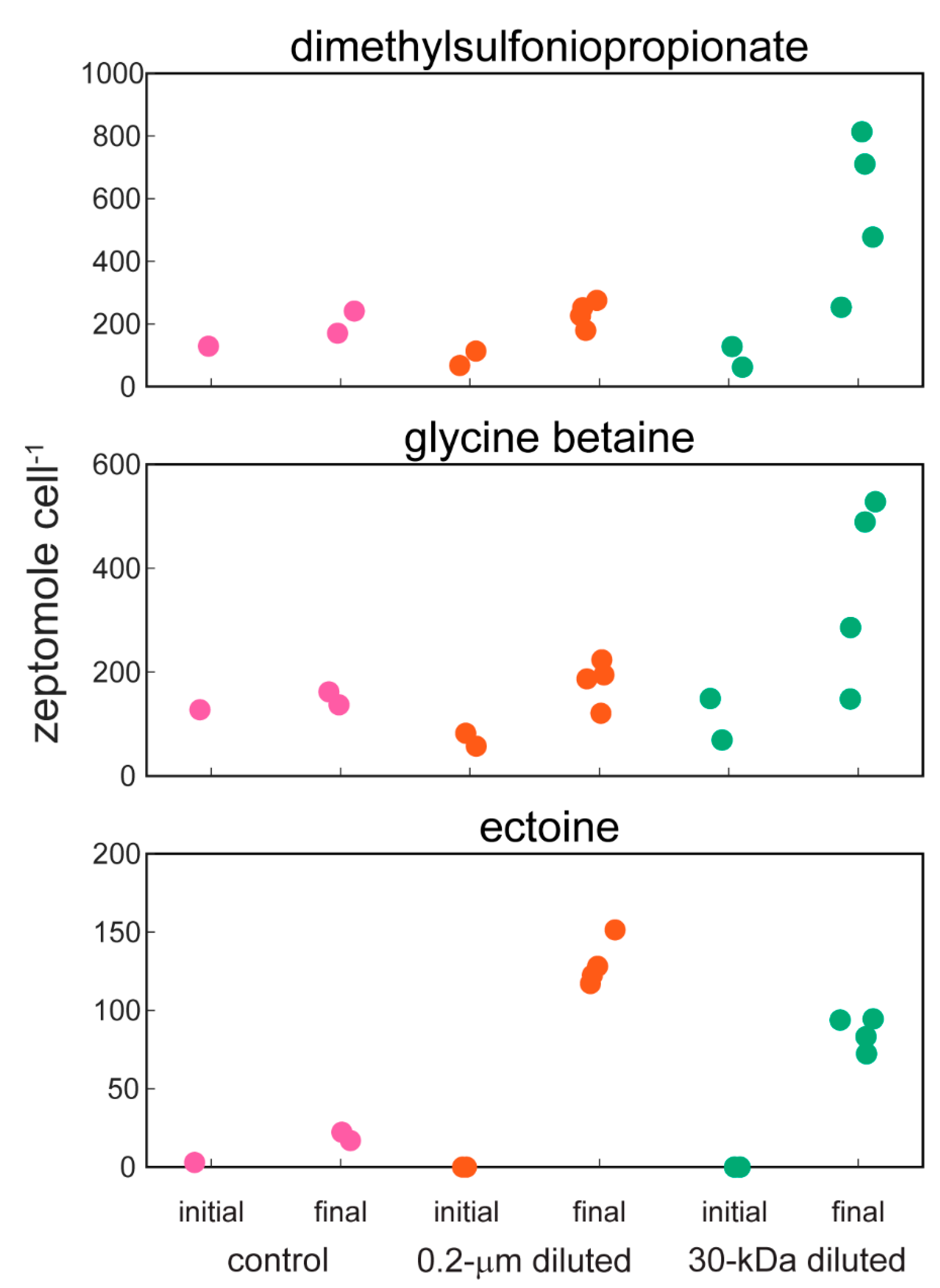
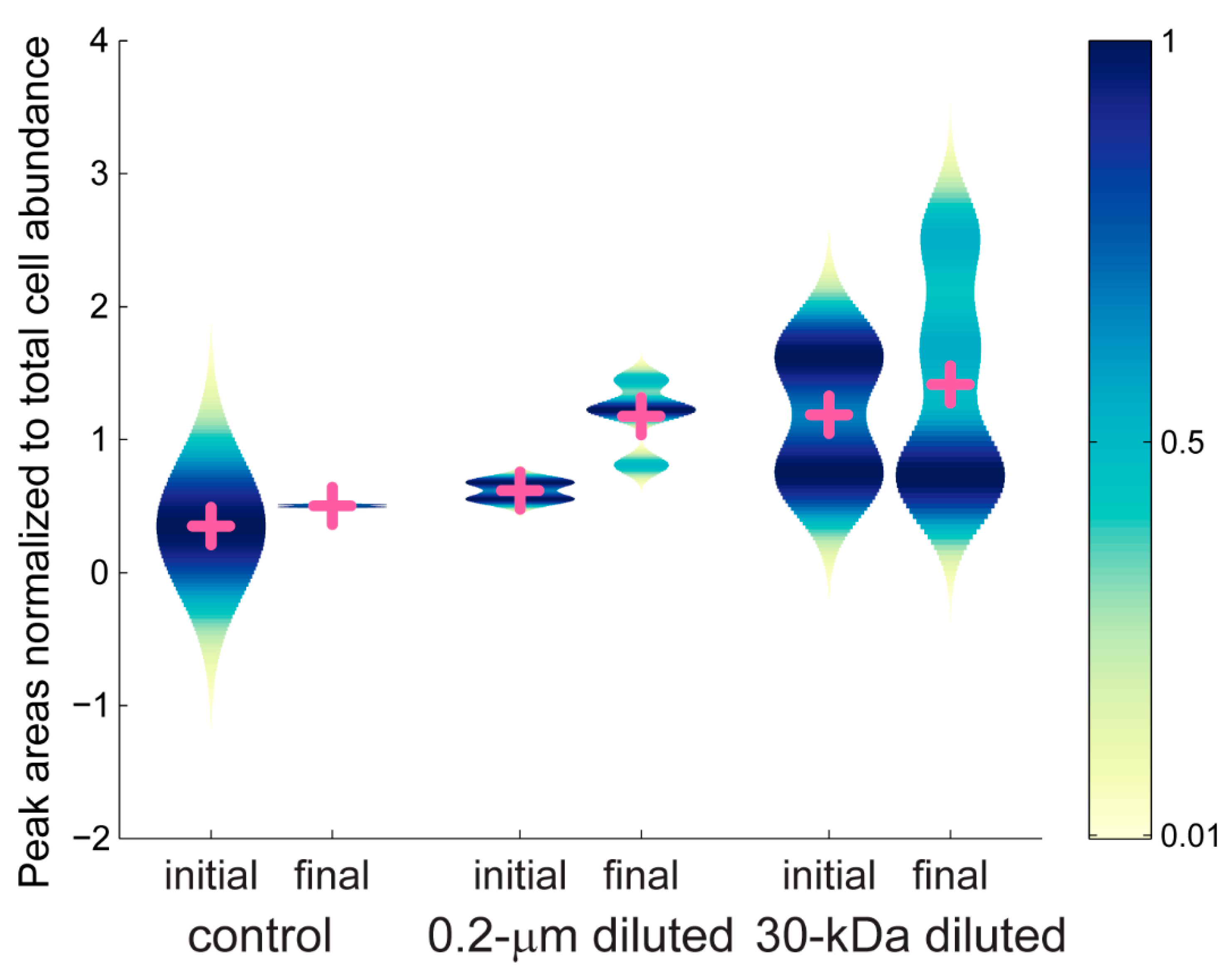
2.2. Targeted Metabolomics Data Results
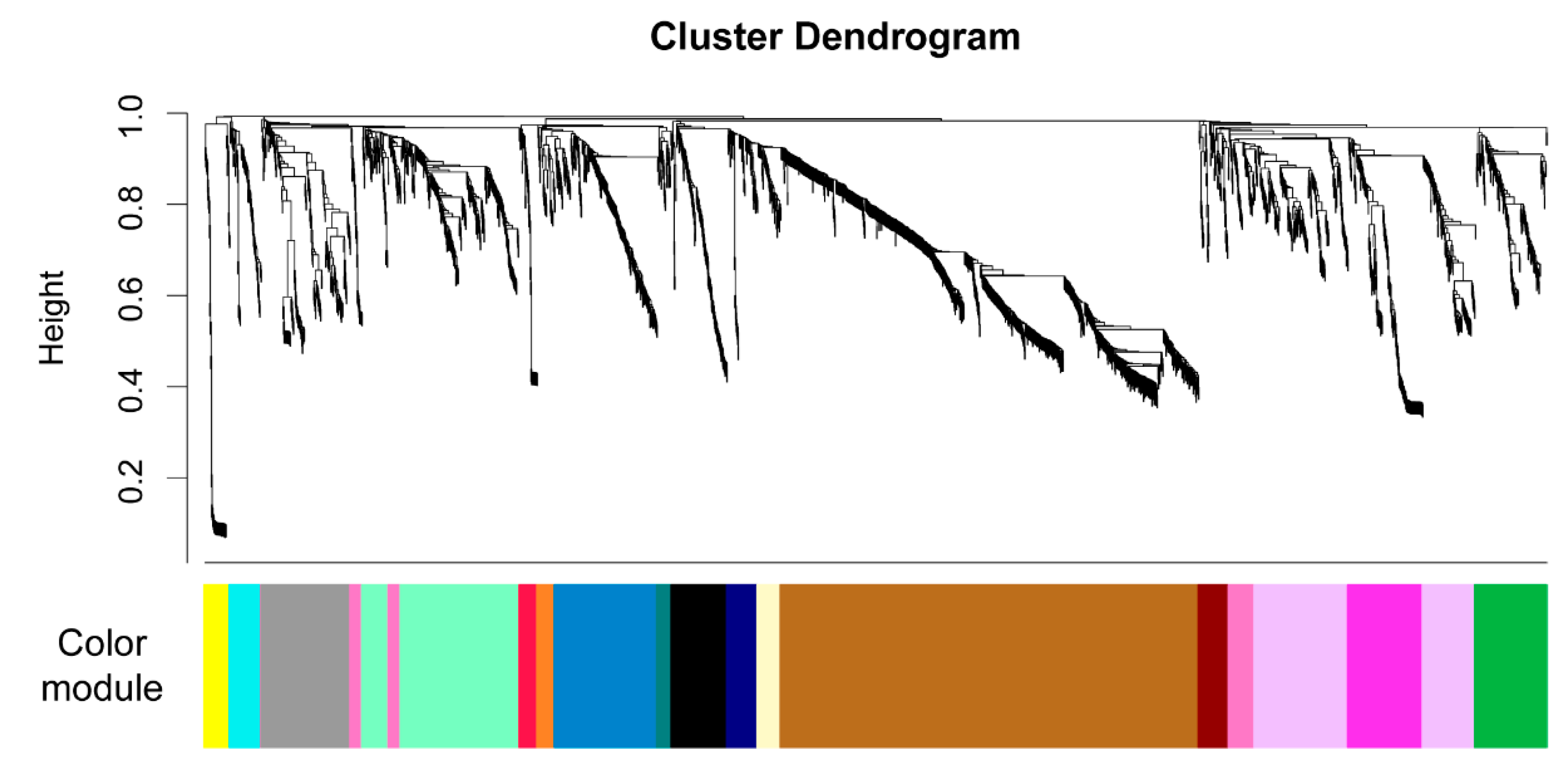
2.3. Untargeted Metabolomics Data from LC FT-ICR MS Analysis
3. Discussion
4. Materials and Methods
4.1. Experimental Setup
4.2. Abundances of Microbial-Sized Cells
4.3. Concentration of Total Organic Carbon (TOC)
4.4. Processing of Seawater Samples for Intracellular Metabolomics
4.5. Targeted and Untargeted Mass Spectrometry
4.6. Statistics and Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fiore, C.L.; Longnecker, K.; Kido Soule, M.C.; Kujawinski, E.B. Release of ecologically relevant metabolites by the cyanobacterium, Synechococcus elongatus CCMP 1631. Environ. Microbiol. 2015, 17, 3949–3963. [Google Scholar] [CrossRef]
- Baran, R.; Bowen, B.P.; Bouskill, N.J.; Brodie, E.L.; Yannone, S.M.; Northen, T.R. Metabolite identification in Synechococcus sp. PCC 7002 using untargeted stable isotope assisted metabolite profiling. Anal. Chem. 2010, 82, 9034–9042. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Dittmar, T.; Bondarev, V.; Weber, R.J.M.; Viant, M.R.; Schulz-Vogt, H.N. Exo-metabolome of Pseudovibrio sp. FO-BEG1 analyzed by ultra-high resolution mass spectrometry and the effect of phosphate limitation. PLoS ONE 2014, 9, e96038. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Kido Soule, M.C.; Kujawinski, E.B. Evidence for quorum sensing and differential metabolite production by a marine bacterium in response to DMSP. ISME J. 2016, 10, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.A.; Giovannoni, S.J.; Hansell, D.A.; Stuart, J.G.; Parsons, R.; Vergin, K. Interactions among dissolved organic carbon, microbial processes, and community structure in the mesopelagic zone of the northwestern Sargasso Sea. Limnol. Oceanogr. 2004, 49, 1073–1083. [Google Scholar] [CrossRef]
- Pomeroy, L.R.; Sheldon, J.E.; Sheldon, W.M., Jr. Changes in bacterial numbers and leucine assimilation during estimations of microbial respiratory rates in seawater by the precision Winkler method. Appl. Environ. Microbiol. 1994, 60, 328–332. [Google Scholar] [CrossRef]
- Longnecker, K.; Wilson, M.J.; Sherr, E.B.; Sherr, B.F. Effect of top-down control on cell-specific activity and diversity of active marine bacterioplankton. Aquat. Microb. Ecol. 2010, 58, 153–165. [Google Scholar] [CrossRef]
- Kido Soule, M.C.; Longnecker, K.; Johnson, W.M.; Kujawinski, E.B. Environmental metabolomics: Analytical strategies. Mar. Chem. 2015, 177, 374–387. [Google Scholar] [CrossRef]
- Landry, M.R.; Hassett, R.P. Estimating the grazing impact of marine micro-zooplankton. Mar. Biol. 1982, 67, 283–288. [Google Scholar] [CrossRef]
- Evans, C.; Archer, S.D.; Jacquet, S.; Wilson, W.H. Direct estimates of the contribution of viral lysis and microzooplankton grazing to the decline of a Micromonas spp. population. Aquat. Microb. Ecol. 2003, 30, 207–219. [Google Scholar] [CrossRef]
- Agis, M.; Granda, A.; Dolan, J.R. A cautionary note: Examples of possible microbial community dynamics in dilution grazing experiments. J. Exp. Mar. Biol. Ecol. 2007, 341, 176–183. [Google Scholar] [CrossRef]
- Dolan, J.R.; McKeon, K. The reliability of grazing rate estimates from dilution experiments: Have we over-estimated rates of organic carbon consumption by microzooplankton? Ocean. Sci. 2005, 1, 1–7. [Google Scholar] [CrossRef]
- Calbet, A.; Saiz, E. Effects of trophic cascades in dilution grazing experiments: From artificial saturated feeding responses to positive slopes. J. Plankton Res. 2013, 35, 1183–1191. [Google Scholar] [CrossRef]
- Bennette, N.B.; Eng, J.F.; Dismukes, G.C. An LC-MS-based chemical and analytical method for targeted metabolite quantification in the model cyanobacterium Synechococcus sp. PCC 7002. Anal. Chem. 2011, 83, 3808–3816. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef]
- DiLeo, M.V.; Strahan, G.D.; den Bakker, M.; Hoekenga, O.A. Weighted Correlation Network Analysis (WGCNA) applied to the tomato fruit metabolome. PLoS ONE 2011, 6, e26683. [Google Scholar] [CrossRef]
- Lusczek, E.R.; Lexcen, D.R.; Witowski, N.E.; Mulier, K.E.; Beilman, G. Urinary metabolic network analysis in trauma, hemorrhagic shock, and resuscitation. Metabolomics 2013, 9, 223–235. [Google Scholar] [CrossRef]
- Sherr, E.B.; Sherr, B.F.; Sigmon, C.T. Activity of marine bacteria under incubated and in situ conditions. Aquat. Microb. Ecol. 1999, 20, 213–223. [Google Scholar] [CrossRef]
- Sieracki, M.E.; Cucci, T.L.; Nicinski, J. Flow cytometric analysis of 5-cyano-2,3-ditolyl tetrazolium chloride activity of marine bacterioplankton in dilution cultures. Appl. Environ. Microbiol. 1999, 65, 2409–2417. [Google Scholar] [CrossRef]
- Pree, B.; Kuhlisch, C.; Pohnert, G.; Sazhin, A.F.; Jakobsen, H.H.; Paulsen, M.L.; Frischer, M.E.; Stoecker, D.; Nejstgaard, J.C.; Larsen, A. A simple adjustment to test reliability of bacterivory rates derived from the dilution method. Limnol. Oceanogr. Meth. 2016, 14, 114–123. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Bell, T.M. Biological considerations in the measurement of dissolved free amino acids in seawater and implications for chemical and microbiological studies. Mar. Ecol. Prog. Ser. 1985, 25, 13–21. [Google Scholar] [CrossRef]
- Pasulka, A.L.; Samo, T.J.; Landry, M.R. Grazer and viral impacts on microbial growth and mortality in the southern California Current Ecosystem. J. Plankton Res. 2015, 37, 320–336. [Google Scholar] [CrossRef]
- Calvo-Diaz, A.; Diaz-Perez, L.; Suarez, L.A.; Moran, X.A.G.; Teira, E.; Maranon, E. Decrease in the autotrophic-to-heterotrophic biomass ratio of picoplankton in oligotrophic marine waters due to bottle enclosure. Appl. Environ. Microbiol. 2011, 77, 5739–5746. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.T. Effect of protistan bacterivory on coastal bacterioplankton diversity. Aquat. Microb. Ecol. 1999, 20, 261–272. [Google Scholar] [CrossRef]
- Cram, J.A.; Parada, A.E.; Fuhrman, J.A. Dilution reveals how viral lysis and grazing shape microbial communities. Limnol. Oceanogr. 2016, 61, 889–905. [Google Scholar] [CrossRef]
- Hagström, A.; Ammerman, J.W.; Henrichs, S.; Azam, F. Bacterioplankton growth in seawater: II. Organic matter utilization during steady-state growth in seawater cultures. Mar. Ecol. Prog. Ser. 1984, 18, 41–48. [Google Scholar] [CrossRef]
- Ducklow, H. Bacterial production and biomass in the oceans. In Microbial Ecology of the Oceans; Kirchman, D.L., Ed.; Wiley-Liss, Inc.: New York, NY, USA, 2000; pp. 85–120. [Google Scholar]
- Chin-Leo, G.; Kirchman, D.L. Unbalanced growth in natural assemblages of marine bacterioplankton. Mar. Ecol. Prog. Ser. 1990, 63, 1–8. [Google Scholar] [CrossRef]
- Shiah, F.K.; Ducklow, H.W. Bacterioplankton growth responses to temperature and chlorophyll variations in estuaries measured by thymidine:leucine incorporation ratio. Aquat. Microb. Ecol. 1997, 13, 151–159. [Google Scholar] [CrossRef][Green Version]
- Longnecker, K.; Sherr, B.F.; Sherr, E.B. Variation in cell-specific rates of leucine and thymidine incorporation by high and low nucleic acid content marine bacteria off the Oregon coast. Aquat. Microb. Ecol. 2006, 43, 113–125. [Google Scholar] [CrossRef]
- Kurz, M. Compatible solute influence on nucleic acids: Many questions but few answers. Saline Systems 2008, 4, 1–14. [Google Scholar] [CrossRef]
- Roychoudhury, A.; Haussinger, D.; Oesterhelt, F. Effect of the compatible solute ectoine on the stability of the membrane proteins. Protein Peptide Lett. 2012, 19, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Harishchandra, R.K.; Wulff, S.; Lentzen, G.; Neuhaus, T.; Galla, H.-J. The effect of compatible solute ectoines on the structural organization of lipid monolayer and bilayer membranes. Biophys. Chem. 2010, 150, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Hoehler, T.M.; Jørgensen, B.B. Microbial life under extreme energy limitation. Nat. Rev. Microbiol. 2013, 11, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.A.; del Giorgio, P.A.; Herndl, G.J. Microbes and the dissipation of energy and respiration: From cells to ecosystems. Oceanography 2007, 20, 62–73. [Google Scholar] [CrossRef]
- Lipson, D.A. The complex relationship between microbial growth rate and yield and its implications for ecosystem processes. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Roberts, M.F. Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Syst. 2005, 1, 5. [Google Scholar] [CrossRef]
- Gifford, S.M.; Sharma, S.; Booth, M.; Moran, M.A. Expression patterns reveal niche diversification in a marine microbial assemblage. ISME J. 2013, 7, 281–298. [Google Scholar] [CrossRef]
- Johnson, W.M.; Longnecker, K.; Kido Soule, M.C.; Arnold, W.A.; Bhatia, M.P.; Hallam, S.J.; Van Mooy, B.A.S.; Kujawinski, E.B. Metabolite composition of sinking particles differs from surface suspended particles across a latitudinal transect in the South Atlantic. Limnol. Oceanogr. 2020, 65, 111–127. [Google Scholar] [CrossRef]
- Sherr, E.B.; Sherr, B.F.; Wheeler, P.A. Distribution of coccoid cyanobacteria and small eukaryotic phytoplankton in the upwelling ecosystem off the Oregon coast during 2001 and 2002. Deep-Sea Res. Pt. II 2005, 52, 317–330. [Google Scholar] [CrossRef]
- Marie, D.; Partensky, F.; Jacquet, S.; Vaulot, D. Enumeration and cell cycle analysis of natural populations of marine picoplankton by flow cytometry using the nucleic acid stain SYBR Green I. Appl. Environ. Microbiol. 1997, 63, 186–193. [Google Scholar] [CrossRef]
- Sherr, E.B.; Caron, D.A.; Sherr, B.F. Staining of heterotrophic protists for visualization via epifluorescence microscopy. In Handbook of Methods in Aquatic Microbial Ecology; Kemp, P.F., Sherr, B.F., Sherr, E.B., Cole, J.J., Eds.; Lewis Publishers: Boca Raton, FL, USA, 1993; pp. 213–228. [Google Scholar]
- Rabinowitz, J.D.; Kimball, E. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 2007, 79, 6167–6173. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Melamud, E.; Vastag, L.; Rabinowitz, J.D. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 2010, 82, 9818–9826. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Kido Soule, M.C.; Kujawinski, E.B. Interpreting the impact of matrix on extraction efficiency and instrument response in a targeted metabolomics method. Limnol. Oceanogr. Meth. 2017, 15, 417–428. [Google Scholar] [CrossRef]
- Dittmar, T.; Koch, B.; Hertkorn, N.; Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr. Meth. 2008, 6, 230–235. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Bottcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Longnecker, K.; Futrelle, J.; Coburn, E.; Kido Soule, M.C.; Kujawinski, E.B. Environmental metabolomics: Databases and tools for data analysis. Mar. Chem. 2015, 177, 366–373. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | % Whole Seawater | Time Point (# of Samples) | TOC (µM) |
|---|---|---|---|
| control | 100% | initial (n = 1) final (n = 2) | 76.9 80.3, 81.0 |
| 0.2-µm diluted | 45% | initial (n = 1) final (n = 2) | 82.0 77.5, 81.8 |
| 0.2-µm diluted | 20% | initial (n = 1) final (n = 2) | * 84.7, 80.0 |
| 30-kDa diluted | 45% | initial (n = 1) final (n = 2) | 75.6 69.0, 80.8 |
| 30-kDa diluted | 20% | initial (n = 1) final (n = 2) | 81.7 77.7, 86.0 |
| Metabolite | p-Value | 0.2-µm Diluted (+/-) | 30-kDa Diluted (+/-) |
|---|---|---|---|
| guanine | 0.0002 | x (-) | x (-) |
| glutamic acid | 0.0263 | x (+) | |
| phenylalanine | 0.0105 | x (+) | |
| ectoine | 0.0001 | x (+) | x (+) |
| Color (Arbitrary) | # mzRT Features | Positive Correlation | Negative Correlation |
|---|---|---|---|
| blue | 218 | ectoine proline | ciliatine guanine |
| magenta | 159 | glutamic acid | |
| green | 153 | xanthosine | glutamine |
| navy | 65 | D-ribose 5-phosphate syringic acid | |
| maroon | 64 | inosine 5’-monophosphate | |
| yellow | 54 | 3-mercaptopropionic acid | |
| beige | 49 | NAD 1 pantothenic acid | |
| red | 38 | proline | inosine |
| orange | 37 | glutamic acid phenylalanine | ciliatine guanine |
| brown | 890 | ||
| lavender | 310 | ||
| mint | 309 | ||
| grey | 189 | ||
| black | 118 | ||
| pink | 107 | ||
| cyan | 67 | ||
| teal | 31 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longnecker, K.; Kujawinski, E.B. Intracellular Metabolites in Marine Microorganisms during an Experiment Evaluating Microbial Mortality. Metabolites 2020, 10, 105. https://doi.org/10.3390/metabo10030105
Longnecker K, Kujawinski EB. Intracellular Metabolites in Marine Microorganisms during an Experiment Evaluating Microbial Mortality. Metabolites. 2020; 10(3):105. https://doi.org/10.3390/metabo10030105
Chicago/Turabian StyleLongnecker, Krista, and Elizabeth B. Kujawinski. 2020. "Intracellular Metabolites in Marine Microorganisms during an Experiment Evaluating Microbial Mortality" Metabolites 10, no. 3: 105. https://doi.org/10.3390/metabo10030105
APA StyleLongnecker, K., & Kujawinski, E. B. (2020). Intracellular Metabolites in Marine Microorganisms during an Experiment Evaluating Microbial Mortality. Metabolites, 10(3), 105. https://doi.org/10.3390/metabo10030105