Evaluation of Different Tandem MS Acquisition Modes to Support Metabolite Annotation in Human Plasma Using Ultra High-Performance Liquid Chromatography High-Resolution Mass Spectrometry for Untargeted Metabolomics

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
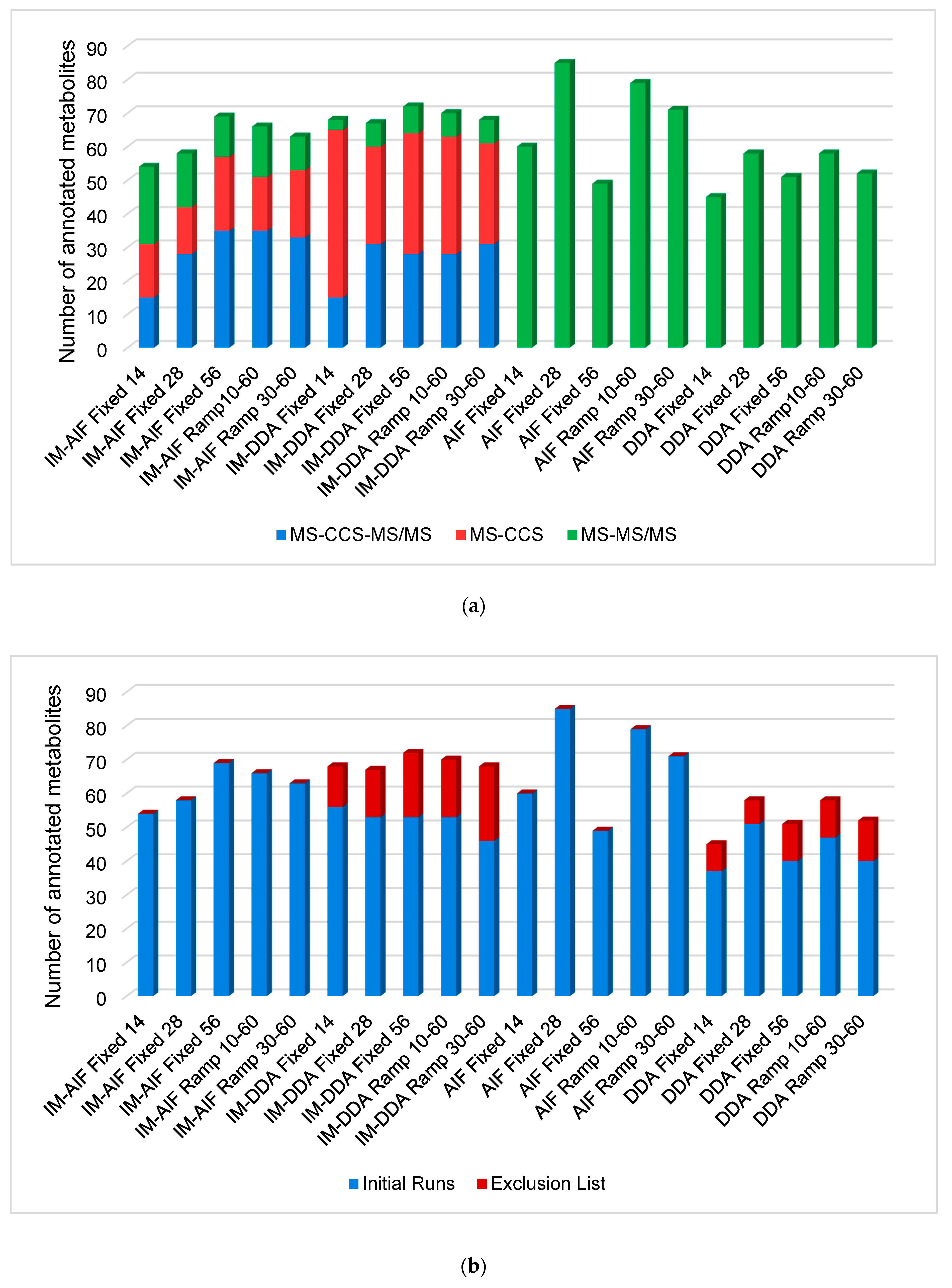
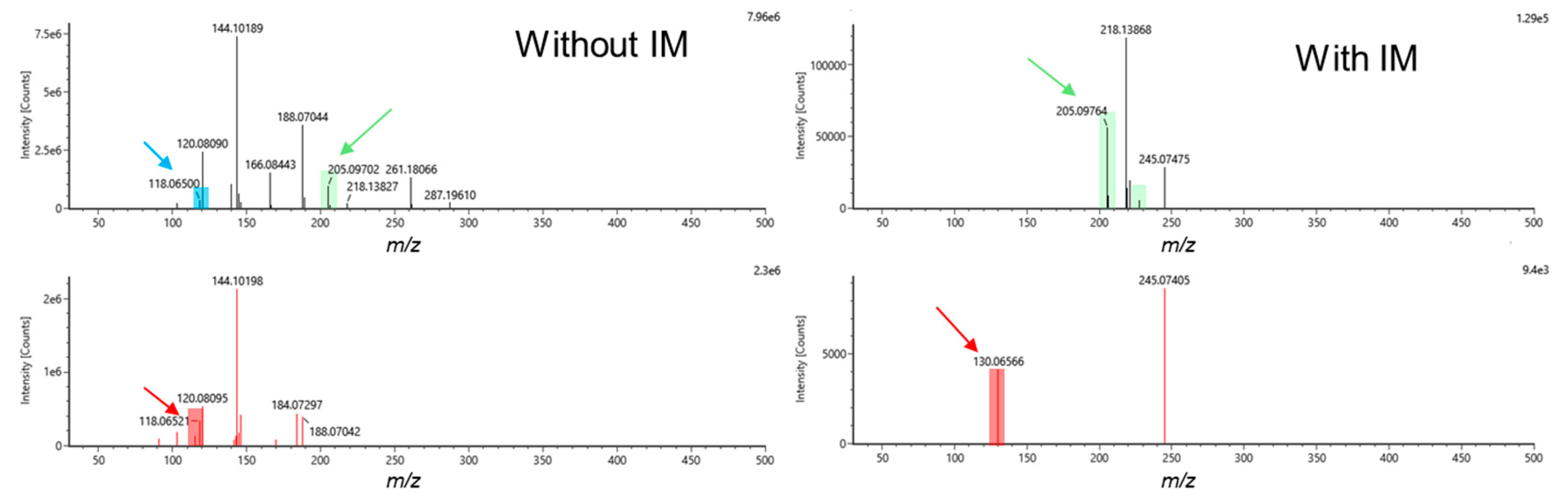
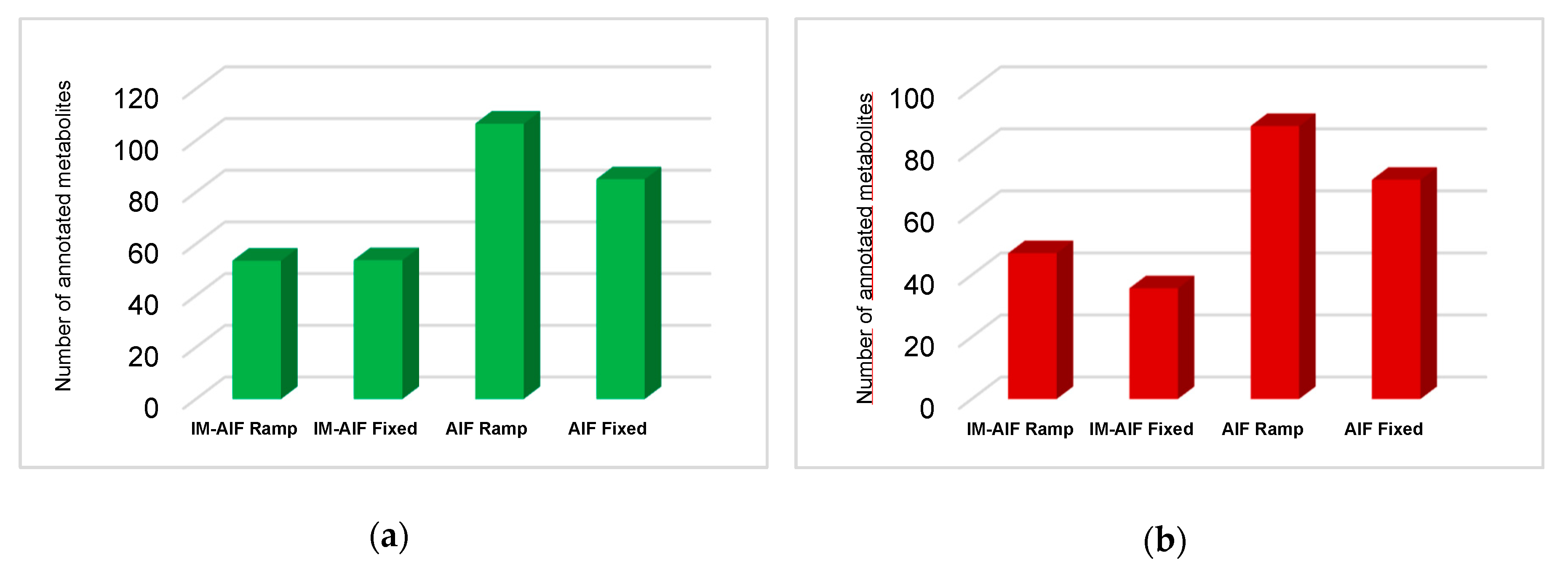
2.1. Investigation of All Tandem MS Modes under RPLC Conditions
2.2. Evaluating the Best Tandem MS Modes for HILIC Separations
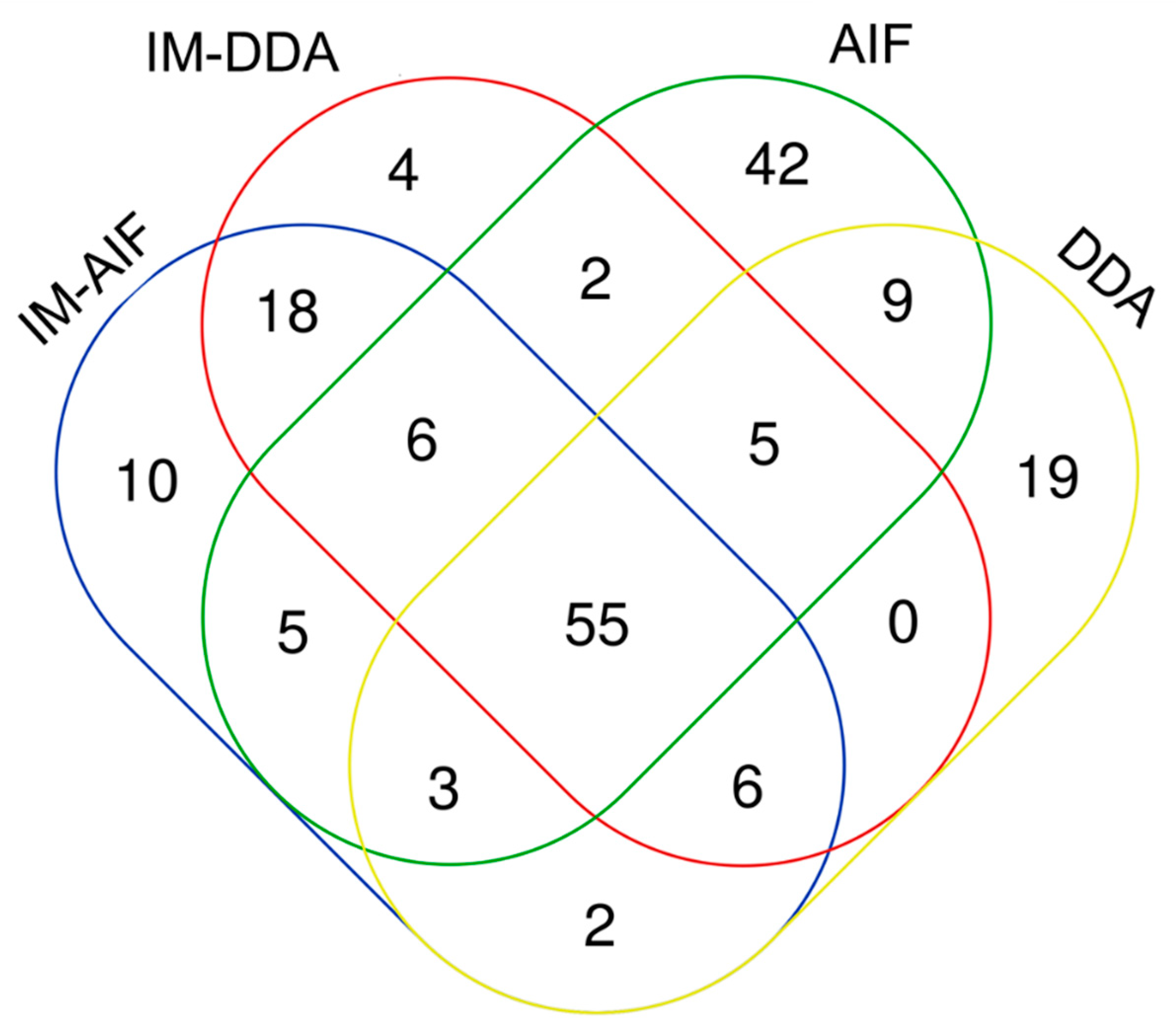
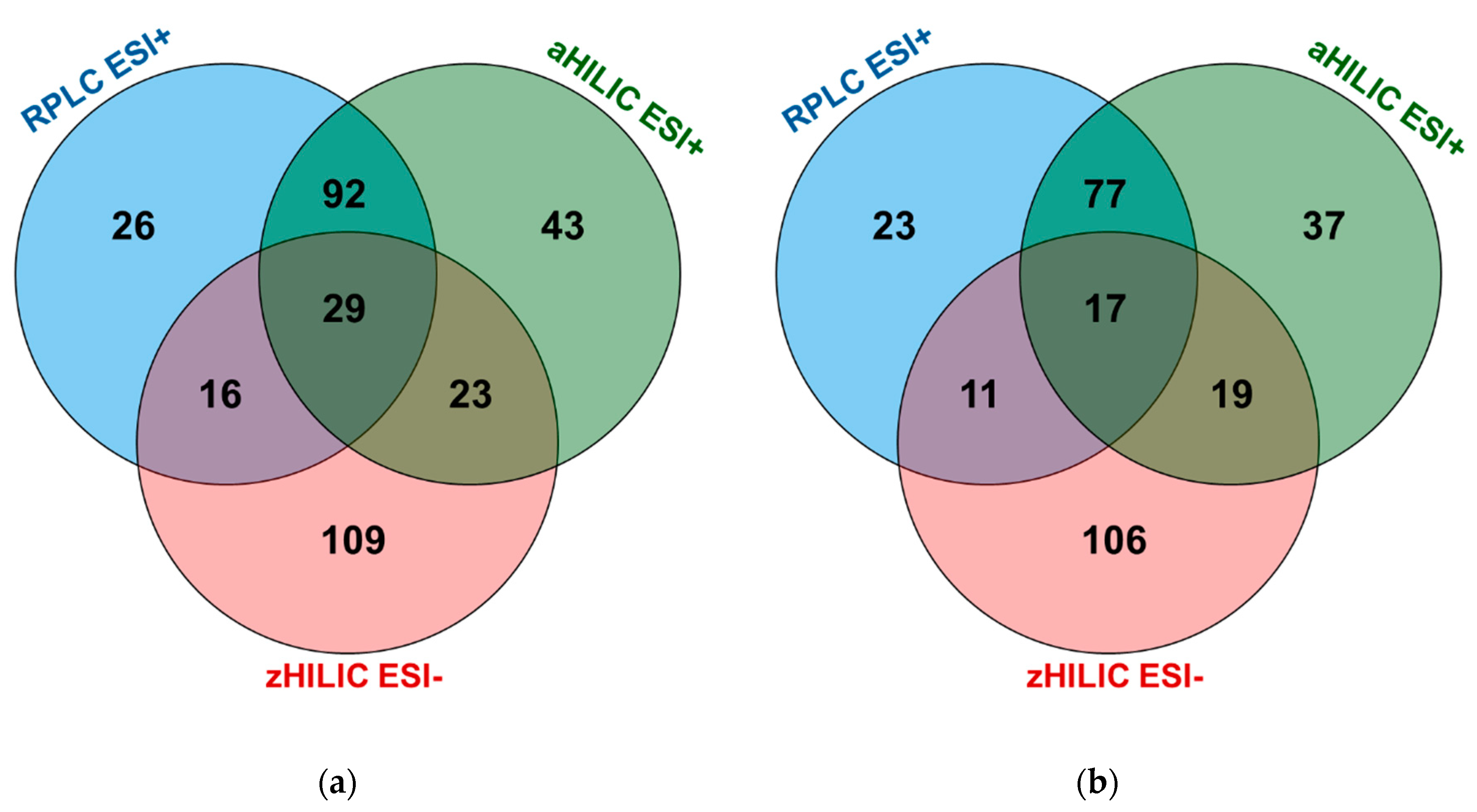
2.3. Optimal Combination of LC and MS/MS Modes to Improve Human Plasma Metabolome Coverage
3. Materials and Methods
3.1. Chemicals
3.2. Human Plasma
3.3. Sample Preparation
3.4. Liquid Chromatographic Conditions
3.5. Mass Spectrometric Conditions
3.6. Exclusion Lists
3.7. Analytical Sequence
- i.
- Condition the LC to the gradient mode.
- ii.
- Evaluate the performance of the analytical platform [31].
- iii.
- Condition and stabilize the LC and HRMS systems to the plasma matrix.
- iv.
- Use the exact same matrix sample as the following runs of interest.
3.8. Analysis of Raw Data and Metabolite Annotation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 1–17. [Google Scholar] [CrossRef]
- Gagnebin, Y.; Julien, B.; Belén, P.; Serge, R. Metabolomics in chronic kidney disease: Strategies for extended metabolome coverage. J. Pharm. Biomed. Anal. 2018, 161, 313–325. [Google Scholar] [CrossRef]
- Sévin, D.C.; Kuehne, A.; Zamboni, N.; Sauer, U. Biological insights through nontargeted metabolomics. Curr. Opin. Biotechnol. 2015, 34, 1–8. [Google Scholar] [CrossRef]
- Jacob, M.; Lopata, A.L.; Dasouki, M.; Rahman, A.M.A. Metabolomics toward personalized medicine. Mass Spectrom. Rev. 2019, 38, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Godzien, J.; Kalaska, B.; Adamska-Patruno, E.; Siroka, J.; Ciborowski, M.; Kretowski, A.; Barbas, C. Oxidized glycerophosphatidylcholines in diabetes through non-targeted metabolomics: Their annotation and biological meaning. J. Chromatogr. B 2019, 1120, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Adamska-Patruno, E.; Samczuk, P.; Ciborowski, M.; Godzien, J.; Pietrowska, K.; Bauer, W.; Gorska, M.; Barbas, C.; Kretowski, A. Metabolomics Reveal Altered Postprandial Lipid Metabolism After a High-Carbohydrate Meal in Men at High Genetic Risk of Diabetes. J. Nutr. 2019, 149, 915–922. [Google Scholar] [CrossRef]
- Armitage, E.G.; Barbas, C. Metabolomics in cancer biomarker discovery: Current trends and future perspectives. J. Pharm. Biomed. Anal. 2014, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, T.; Ciborowski, M.; Kisluk, J.; Kretowski, A.; Barbas, C. Mass spectrometry based proteomics and metabolomics in personalized oncology. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2020, 1866, 165690. [Google Scholar] [CrossRef]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The Human Serum Metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef]
- Fernández-García, M.; Rey-Stolle, F.; Boccard, J.; Reddy, V.P.; García, A.; Cumming, B.M.; Steyn, A.J.C.; Rudaz, S.; Barbas, C. Comprehensive Examination of the Mouse Lung Metabolome Following Mycobacterium tuberculosis Infection Using a Multiplatform Mass Spectrometry Approach. J. Proteome Res. 2020, 19, 2053–2070. [Google Scholar] [CrossRef]
- Desfontaine, V.; Losacco, G.L.; Gagnebin, Y.; Pezzatti, J.; Farrell, W.P.; González-Ruiz, V.; Serge, R.; Veuthey, J.-L.; Guillarme, D. Applicability of supercritical fluid chromatography – mass spectrometry to metabolomics. I – Optimization of separation conditions for the simultaneous analysis of hydrophilic and lipophilic substances. J. Chromatogr. A 2018, 1562, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Drouin, N.; Pezzatti, J.; Gagnebin, Y.; González-Ruiz, V.; Schappler, J.; Rudaz, S. Effective mobility as a robust criterion for compound annotation and identification in metabolomics: Toward a mobility-based library. Anal. Chim. Acta 2018, 1032, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Wernisch, S.; Pennathur, S. Evaluation of coverage, retention patterns, and selectivity of seven liquid chromatographic methods for metabolomics. Anal. Bioanal. Chem. 2016, 408, 6079–6091. [Google Scholar] [CrossRef] [PubMed]
- Gallart-Ayala, H.; Konz, I.; Mehl, F.; Teav, T.; Oikonomidi, A.; Peyratout, G.; Van Der Velpen, V.; Popp, J.; Ivanisevic, J. A global HILIC-MS approach to measure polar human cerebrospinal fluid metabolome: Exploring gender-associated variation in a cohort of elderly cognitively healthy subjects. Anal. Chim. Acta 2018, 1037, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 1–12. [Google Scholar] [CrossRef]
- Pezzatti, J.; González-Ruiz, V.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Guillarme, D.; Schappler, J.; Picard, D.; Boccard, J.; Serge, R. A scoring approach for multi-platform acquisition in metabolomics. J. Chromatogr. A 2019, 1592, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Narduzzi, L.; Royer, A.-L.; Bichon, E.; Guitton, Y.; Buisson, C.; Le Bizec, B.; Dervilly-Pinel, G. Ammonium Fluoride as Suitable Additive for HILIC-Based LC-HRMS Metabolomics. Metabolites 2019, 9, 292. [Google Scholar] [CrossRef]
- Randazzo, G.M.; Tonoli, D.; Hambye, S.; Guillarme, D.; Jeanneret, F.; Nurisso, A.; Goracci, L.; Julien, B.; Rudaz, S. Prediction of retention time in reversed-phase liquid chromatography as a tool for steroid identification. Anal. Chim. Acta 2016, 916, 8–16. [Google Scholar] [CrossRef]
- Fenaille, F.; Saint-Hilaire, P.B.; Rousseau, K.; Junot, C. Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? J. Chromatogr. A 2017, 1526, 1–12. [Google Scholar] [CrossRef]
- Gagnebin, Y.; Pezzatti, J.; Lescuyer, P.; Boccard, J.; Ponte, B.; Serge, R. Toward a better understanding of chronic kidney disease with complementary chromatographic methods hyphenated with mass spectrometry for improved polar metabolome coverage. J. Chromatogr. B 2019, 1116, 9–18. [Google Scholar] [CrossRef]
- Gagnebin, Y.; Pezzatti, J.; Lescuyer, P.; Boccard, J.; Ponte, B.; Rudaz, S. Combining the advantages of multilevel and orthogonal partial least squares data analysis for longitudinal metabolomics: Application to kidney transplantation. Anal. Chim. Acta 2019, 1099, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Blaženović, I.; Kind, T.; Sa, M.R.; Ji, J.; Vaniya, A.; Wancewicz, B.; Roberts, B.S.; Torbašinović, H.; Lee, T.; Mehta, S.S.; et al. Structure Annotation of All Mass Spectra in Untargeted Metabolomics. Anal. Chem. 2019, 91, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Blaženović, I.; Kind, T.; Ji, J.; Fiehn, O. Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Ivanisevic, J.; Thomas, A.; Sidibé, J.; Teav, T.; Guijas, C.; Aisporna, A.E.; Rinehart, D.; Hoang, L.; et al. XCMS-MRM and METLIN-MRM: A cloud library and public resource for targeted analysis of small molecules. Nat. Methods 2018, 15, 681–684. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; Le Bizec, B.; Monteau, F.; García-Campaña, A.M.; Dervilly-Pinel, G. Collision Cross Section (CCS) Database: An Additional Measure to Characterize Steroids. Anal. Chem. 2018, 90, 4616–4625. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; D’Atri, V.; Barknowitz, G.; Fanuel, M.; Pezzatti, J.; Dreolin, N.; Ropartz, D.; Monteau, F.; Vigneau, E.; Rudaz, S.; et al. Interlaboratory and Interplatform Study of Steroids Collision Cross Section by Traveling Wave Ion Mobility Spectrometry. Anal. Chem. 2020, 92, 5013–5022. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Allison, P.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Smith, C.A.; Maille, G.O.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. ETLIN: A Metabolite Mass Spectral Database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Gil-De-La-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802. [Google Scholar] [CrossRef]
- Pezzatti, J.; Boccard, J.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Picard, D.; González-Ruiz, V.; Rudaz, S. Implementation of liquid chromatography–high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial. Anal. Chim. Acta 2020, 1105, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Gallart-Ayala, H.; Reinke, S.N.; Mathon, C.; Blankley, R.; Chaleckis, R.; Wheelock, C.E. Development of a Liquid Chromatography–High Resolution Mass Spectrometry Metabolomics Method with High Specificity for Metabolite Identification Using All Ion Fragmentation Acquisition. Anal. Chem. 2017, 89, 7933–7942. [Google Scholar] [CrossRef] [PubMed]
- Plumb, R.S.; Johnson, K.A.; Rainville, P.; Smith, B.W.; Wilson, I.D.; Castro-Perez, J.M.; Nicholson, J.K. UPLC/MSE; a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun. Mass Spectrom. 2006, 20, 1989–1994. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, Y.; Subramanian, R. Comparison of Information-Dependent Acquisition, SWATH, and MSAll Techniques in Metabolite Identification Study Employing Ultrahigh-Performance Liquid Chromatography–Quadrupole Time-of-Flight Mass Spectrometry. Anal. Chem. 2014, 86, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Nash, W.J.; Dunn, W.B. From mass to metabolite in human untargeted metabolomics: Recent advances in annotation of metabolites applying liquid chromatography-mass spectrometry data. TrAC Trends Anal. Chem. 2019, 120, 115324. [Google Scholar] [CrossRef]
- Paglia, G.; Williams, J.P.; Menikarachchi, L.; Thompson, J.W.; Tyldesley-Worster, R.; Halldórsson, S.; Rolfsson, O.; Moseley, A.; Grant, D.; Langridge, J.; et al. Ion Mobility Derived Collision Cross Sections to Support Metabolomics Applications. Anal. Chem. 2014, 86, 3985–3993. [Google Scholar] [CrossRef]
- Ma, X.; Liu, J.; Zhang, Z.; Bo, T.; Bai, Y.; Liu, H. Drift tube ion mobility and four-dimensional molecular feature extraction enable data-independent tandem mass spectrometric ‘omics’ analysis without quadrupole selection. Rapid Commun. Mass Spectrom. 2016, 31, 33–38. [Google Scholar] [CrossRef][Green Version]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Practical application of in silico fragmentation based residue screening with ion mobility high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2017, 31, 1147–1157. [Google Scholar] [CrossRef]
- D’Atri, V.; Causon, T.; Hernandez-Alba, O.; Mutabazi, A.; Veuthey, J.-L.; Cianférani, S.; Guillarme, D. Adding a new separation dimension to MS and LC-MS: What is the utility of ion mobility spectrometry? J. Sep. Sci. 2017, 41, 20–67. [Google Scholar] [CrossRef]
- Mollerup, C.B.; Mardal, M.; Dalsgaard, P.W.; Linnet, K.; Barron, L.P. Prediction of collision cross section and retention time for broad scope screening in gradient reversed-phase liquid chromatography-ion mobility-high resolution accurate mass spectrometry. J. Chromatogr. A 2018, 1542, 82–88. [Google Scholar] [CrossRef]
- Blaženović, I.; Shen, T.; Mehta, S.S.; Kind, T.; Ji, J.; Piparo, M.; Cacciola, F.; Mondello, L.; Fiehn, O. Increasing Compound Identification Rates in Untargeted Lipidomics Research with Liquid Chromatography Drift Time–Ion Mobility Mass Spectrometry. Anal. Chem. 2018, 90, 10758–10764. [Google Scholar] [CrossRef] [PubMed]
- Hinz, C.; Liggi, S.; Mocciaro, G.; Jung, S.M.; Induruwa, I.; Pereira, M.C.; Bryant, C.E.; Meckelmann, S.W.; O’Donnell, V.B.; Farndale, R.W.; et al. A Comprehensive UHPLC Ion Mobility Quadrupole Time-of-Flight Method for Profiling and Quantification of Eicosanoids, Other Oxylipins, and Fatty Acids. Anal. Chem. 2019, 91, 8025–8035. [Google Scholar] [CrossRef] [PubMed]
- Mairinger, T.; Causon, T.J.; Hann, S. The potential of ion mobility–mass spectrometry for non-targeted metabolomics. Curr. Opin. Chem. Biol. 2018, 42, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Drouin, N.; Mielcarek, A.; Wenz, C.; Rudaz, S. Evaluation of ion mobility in capillary electrophoresis coupled to mass spectrometry for the identification in metabolomics. Electrophoresis 2020. [Google Scholar] [CrossRef]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef]
- Hilaire, P.B.S.; Rousseau, K.; Seyer, A.; Dechaumet, S.; Damont, A.; Junot, C.; Fenaille, F. Comparative Evaluation of Data Dependent and Data Independent Acquisition Workflows Implemented on an Orbitrap Fusion for Untargeted Metabolomics. Metabolites 2020, 10, 158. [Google Scholar] [CrossRef]
- Kirwan, J.A.; Brennan, L.; Broadhurst, D.; Fiehn, O.; Cascante, M.; Dunn, W.B.; Schmidt, M.A.; Velagapudi, V. Preanalytical Processing and Biobanking Procedures of Biological Samples for Metabolomics Research: A White Paper, Community Perspective (for “Precision Medicine and Pharmacometabolomics Task Group”—The Metabolomics Society Initiative). Clin. Chem. 2018, 64, 1158–1182. [Google Scholar] [CrossRef]
- Mairinger, T.; Kurulugama, R.; Causon, T.J.; Stafford, G.; Fjeldsted, J.; Hann, S. Rapid screening methods for yeast sub-metabolome analysis with a high-resolution ion mobility quadrupole time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom. 2019, 33, 66–74. [Google Scholar] [CrossRef]
- Rochat, B. Proposed Confidence Scale and ID Score in the Identification of Known-Unknown Compounds Using High Resolution MS Data. J. Am. Soc. Mass Spectrom. 2017, 28, 709–723. [Google Scholar] [CrossRef]
- Pezzatti, J.; Bergé, M.; Boccard, J.; Codesido, S.; Gagnebin, Y.; Viollier, P.H.; González-Ruiz, V.; Rudaz, S. Choosing an Optimal Sample Preparation in Caulobacter crescentus for Untargeted Metabolomics Approaches. Metabolites 2019, 9, 193. [Google Scholar] [CrossRef]
- Thompson, J.W.; Adams, K.J.; Adamski, J.; Asad, Y.; Borts, D.; Bowden, J.A.; Byram, G.; Dang, V.D.; Dunn, W.B.; Fernández, F.M.; et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal. Chem. 2019, 91, 14407–14416. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.E.; Scott, D.R. Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Astarita, G. Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nat. Protoc. 2017, 12, 797–813. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acquisition Mode | AIF with IM | DDA with IM | AIF without IM | DDA without IM |
|---|---|---|---|---|
| Ramped | IM-AIF Ramp 10–60 | IM-DDA Ramp 10–60 | AIF Ramp 10–60 | DDA Ramp 10–60 |
| IM-AIF Ramp 30–60 | IM-DDA Ramp 30–60 | AIF Ramp 30–60 | DDA Ramp 30–60 | |
| Fixed | IM-AIF Fixed 14 | IM-DDA Fixed 14 | AIF Fixed 14 | DDA Fixed 14 |
| IM-AIF Fixed 28 | IM-DDA Fixed 28 | AIF Fixed 28 | DDA Fixed 28 | |
| IM-AIF Fixed 56 | IM-DDA Fixed 56 | AIF Fixed 56 | DDA Fixed 56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pezzatti, J.; González-Ruiz, V.; Boccard, J.; Guillarme, D.; Rudaz, S. Evaluation of Different Tandem MS Acquisition Modes to Support Metabolite Annotation in Human Plasma Using Ultra High-Performance Liquid Chromatography High-Resolution Mass Spectrometry for Untargeted Metabolomics. Metabolites 2020, 10, 464. https://doi.org/10.3390/metabo10110464
Pezzatti J, González-Ruiz V, Boccard J, Guillarme D, Rudaz S. Evaluation of Different Tandem MS Acquisition Modes to Support Metabolite Annotation in Human Plasma Using Ultra High-Performance Liquid Chromatography High-Resolution Mass Spectrometry for Untargeted Metabolomics. Metabolites. 2020; 10(11):464. https://doi.org/10.3390/metabo10110464
Chicago/Turabian StylePezzatti, Julian, Víctor González-Ruiz, Julien Boccard, Davy Guillarme, and Serge Rudaz. 2020. "Evaluation of Different Tandem MS Acquisition Modes to Support Metabolite Annotation in Human Plasma Using Ultra High-Performance Liquid Chromatography High-Resolution Mass Spectrometry for Untargeted Metabolomics" Metabolites 10, no. 11: 464. https://doi.org/10.3390/metabo10110464
APA StylePezzatti, J., González-Ruiz, V., Boccard, J., Guillarme, D., & Rudaz, S. (2020). Evaluation of Different Tandem MS Acquisition Modes to Support Metabolite Annotation in Human Plasma Using Ultra High-Performance Liquid Chromatography High-Resolution Mass Spectrometry for Untargeted Metabolomics. Metabolites, 10(11), 464. https://doi.org/10.3390/metabo10110464